Part 3 of the three-part research-use series. Parts 1 and 2 built the framework and walked through BPC-157, TB-500, IGF-1 LR3, and GHK-Cu. This part covers the GH secretagogue class (tesamorelin, CJC-1295 with/without DAC, ipamorelin, MK-677), the risk stratification framework, washout strategy, and the active research on peptides as cancer adjuncts.
The course ends with a closing note that pulls the whole framework together.
Module 13 — Growth Hormone Secretagogues: Same Destination, Different Road
Research and educational purposes only. Not for human consumption.
Why This Module Exists
Growth hormone secretagogues — CJC-1295, ipamorelin, hexarelin, MK-677, GHRP-2, GHRP-6, sermorelin, and others — are some of the most-used peptides in the research community. They raise endogenous IGF-1, which is the same downstream effector that IGF-1 LR3 activates directly.
That raises an obvious question: if elevated IGF-1 is the central cancer-pathway concern, are GH secretagogues just a slower version of the same risk? The answer is nuanced, and the nuance matters.
This module unpacks how endogenous IGF-1 elevation differs from direct LR3 administration, why pulsatile signaling matters biologically, and where the risk picture sits for this entire compound class.
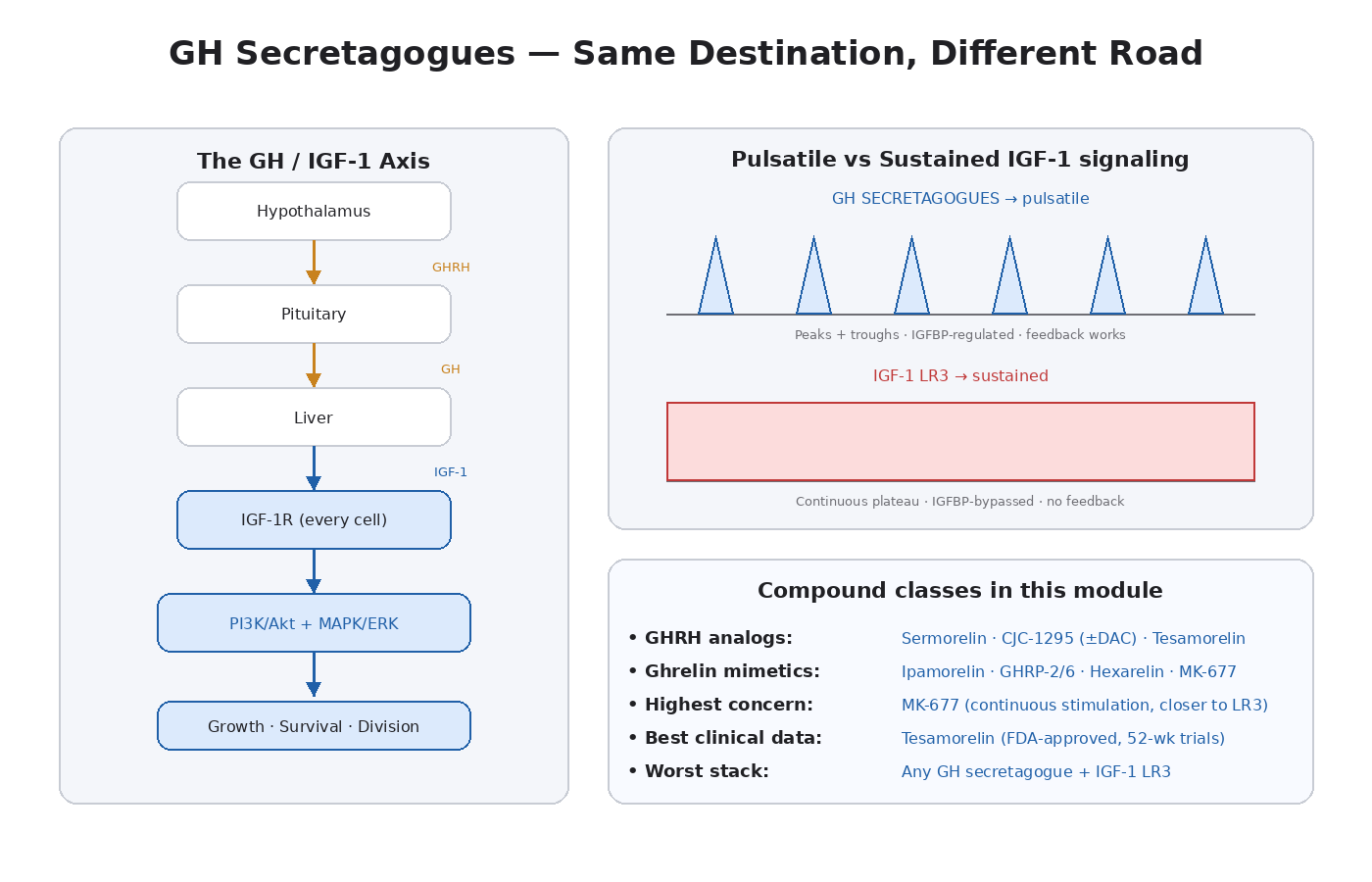
How Growth Hormone Secretagogues Work
The GH/IGF-1 axis is a three-tier signaling cascade:
- Hypothalamus releases growth hormone-releasing hormone (GHRH) and inhibits via somatostatin.
- Pituitary responds to GHRH by releasing growth hormone (GH).
- Liver responds to GH by producing IGF-1.
GH secretagogues intervene at the first two steps.
GHRH Analogs
- Sermorelin — the 1-29 fragment of natural GHRH
- CJC-1295 (without DAC) — modified GHRH with longer half-life (~30 min)
- CJC-1295 (with DAC) — drug affinity complex extension, half-life of ~6–8 days
- Tesamorelin — modified GHRH, FDA-approved for HIV-associated lipodystrophy [15]
These compounds bind the GHRH receptor on the pituitary and stimulate GH release. They mimic natural GHRH signaling.
Ghrelin Mimetics (GHRP-Class)
- Ipamorelin — selective for GH release, minimal cortisol/prolactin effects
- GHRP-2, GHRP-6 — older compounds, stimulate GH release with some appetite/cortisol effects
- Hexarelin — potent GH release, more pronounced cortisol effects
- MK-677 (ibutamoren) — orally active, long half-life, works through the ghrelin receptor (GHSR)
These bind the growth hormone secretagogue receptor (GHSR), a different pituitary receptor than GHRH. They also enhance the amplitude of natural GH pulses.
The two classes are often stacked because their mechanisms are complementary — GHRH analogs and ghrelin mimetics synergize to produce larger GH pulses than either alone.
The Critical Distinction: Pulsatile vs Sustained Signaling
This is the key concept for understanding why GH secretagogues sit differently in the risk framework than direct IGF-1 LR3.
Natural GH secretion is pulsatile. The pituitary releases GH in bursts, mostly during deep sleep but also during exercise and fasting. Between pulses, GH levels are very low. The pulsatile pattern is biologically meaningful — the troughs between pulses allow receptors to reset, prevent chronic downstream signaling, and let various feedback systems operate normally.
When GH pulses hit the liver, the liver produces IGF-1. But there’s a delay — IGF-1 levels respond more slowly and rise and fall over hours rather than minutes. IGF-1 in circulation is largely bound to IGFBPs (especially IGFBP-3) and the acid-labile subunit. Free, biologically active IGF-1 represents a small fraction of total IGF-1.
The result: even under elevated GH secretagogue stimulation, IGF-1 signaling at the receptor level is:
- Modulated by IGFBPs (which are themselves regulated by GH)
- Subject to natural feedback (high IGF-1 inhibits further GH release through the hypothalamus)
- Pulsatile-derived rather than continuously elevated
- Operating through the body’s regulatory infrastructure
Compare to IGF-1 LR3 (Module 11):
- Bypasses IGFBPs entirely
- No natural feedback (LR3 in circulation doesn’t suppress endogenous GH the way native IGF-1 would)
- Continuous high-amplitude signaling for 20+ hours
- Operates outside the regulatory infrastructure
This is biologically a meaningful difference, even though the downstream pathway (IGF-1R → PI3K/Akt/mTOR + MAPK/ERK) is the same.
The Cancer-Pathway Implication
Apply the framework: chronic, IGFBP-bypassing, unregulated IGF-1R signaling is the most concerning configuration. Pulsatile, IGFBP-regulated, feedback-controlled signaling is less concerning.
GH secretagogues sit closer to the second category. They elevate IGF-1, but they do so through the body’s natural infrastructure. IGFBPs continue to modulate signaling. Feedback systems continue to operate. The peaks and troughs of natural physiology are preserved (and enhanced) rather than replaced with constant elevation.
This is not the same as saying GH secretagogues are risk-free. Chronically elevated IGF-1 — even within physiological ranges, just at the upper end — still has the epidemiological cancer signal discussed in Module 11 [11]. The Physicians’ Health Study and similar cohorts measured circulating IGF-1 in people who were producing it endogenously, not injecting LR3.
But the magnitude and chronicity of pathway activation matters, and GH secretagogues produce a fundamentally less aggressive activation profile than LR3.
MK-677: The One With the Highest Concern Within the Class
MK-677 (ibutamoren) deserves a specific call-out because its kinetics differ from the injected GH secretagogues.
MK-677:
- Is orally active
- Has a long half-life (~24 hours)
- Produces continuous (rather than pulsatile) GH stimulation
- Raises IGF-1 to levels comparable with daily GH injection
- Often produces sustained IGF-1 elevation rather than enhanced pulse amplitude
The continuous stimulation profile is closer to LR3 than to the other GH secretagogues. The pulsatile-vs-sustained distinction that protects the other compounds in this class is partially lost with MK-677.
This doesn’t make MK-677 dangerous in the same way as LR3 — it still operates through endogenous IGF-1 with IGFBP modulation — but it’s the compound in this class where the cancer-pathway risk discussion is closest to LR3.
Tesamorelin: The Closest to Clinical Validation
Tesamorelin is the GH secretagogue with the most clinical data. It’s FDA-approved for HIV-associated lipodystrophy and has been studied in trials lasting up to 52 weeks [15].
The cancer-relevant findings:
- No increased cancer signal observed in the clinical trial population over the studied duration
- IGF-1 elevation was modest (within physiological range, not above)
- The pulsatile mechanism was preserved
- Long-term safety beyond ~1 year is still less characterized
This is reassuring data, but it comes with caveats. The trial populations were specific (HIV-associated lipodystrophy patients), the duration was finite, and the doses were specific to that indication. Generalizing to long-term, off-label, recreational research use in a different population requires care.
Where the Risk Profile Sits
Pulling the framework together for this class:
GH secretagogues vs IGF-1 LR3:
- Less aggressive pathway activation (pulsatile vs sustained)
- IGFBP regulation preserved
- Natural feedback systems active
- Lower magnitude of peak signaling
- More physiologically normal pattern
GH secretagogues vs not using any GH/IGF-1 modulator:
- Elevated IGF-1 vs baseline
- Pathway activation that wouldn’t otherwise be occurring
- Epidemiological cancer signal for elevated IGF-1 still applies (within range)
- Chronic use compounds the duration of pathway activation
The middle ground: GH secretagogues are biologically a less aggressive intervention than LR3 but a more aggressive intervention than no intervention. Risk stratification (family history, age, screening, cycle discipline) still applies — just with a milder mechanistic profile to consider.
Practical Implications
Some framework-consistent positions on this class:
Cycling matters. Continuous indefinite use of GH secretagogues produces continuous IGF-1 elevation. Even with the pulsatile signaling advantage, sustained elevation has the same epidemiological signal as natural high-IGF-1 individuals. Cycling restores natural variation.
Dose matters. Microdosing GH secretagogues to nudge IGF-1 slightly above baseline is mechanistically different from pushing IGF-1 well above physiological normal. The risk profile scales with the magnitude of elevation.
Age matters. IGF-1 naturally declines with age. Returning IGF-1 to the level of a 30-year-old in a 60-year-old is a less concerning intervention than pushing a 30-year-old to a level a healthy person would never have. The “what range are you aiming at” question matters.
Stacking matters. Stacking GH secretagogues with LR3 produces the most concerning signaling profile in this entire class (covered in Module 11). Stacking GH secretagogues with each other (CJC + ipamorelin) is the standard pulsatile-enhancing approach and is mechanistically reasonable for short cycles.
Screening matters. As IGF-1 elevation is the central mechanism, knowing baseline IGF-1 and IGFBP-3 levels — and tracking them — is part of responsible research use. Standard cancer screening at age-appropriate intervals also applies.
The Honest Bottom Line
Growth hormone secretagogues activate the same downstream pathway as IGF-1 LR3, but with a fundamentally different signaling kinetic. Pulsatile, IGFBP-regulated, feedback-controlled IGF-1 elevation is biologically less aggressive than direct LR3 administration.
That said, “less aggressive” doesn’t mean “no concern.” The same epidemiological signal that connects elevated IGF-1 to cancer risk operates whether the elevation comes from a pituitary tumor (acromegaly), from natural genetics, or from GH secretagogue administration.
My read: GH secretagogues are a more defensible category than IGF-1 LR3 for someone interested in this kind of research, particularly if used in cycles, at moderate doses, with attention to baseline screening and family history. They are not consequence-free. They are not “safe” in the absolute sense. But the mechanism is meaningfully different from LR3, and the framework should reflect that difference.
MK-677 is the partial exception within the class — its continuous stimulation profile makes it the closest to LR3 in terms of signaling pattern. Tesamorelin is the one with the most reassuring clinical data, with appropriate caveats about study duration and population.
Key Takeaways
- GH secretagogues elevate IGF-1 indirectly through GH-mediated hepatic production.
- GHRH analogs (CJC, sermorelin, tesamorelin) work through one pathway; ghrelin mimetics (ipamorelin, MK-677, hexarelin) through another.
- The class produces pulsatile-pattern IGF-1 elevation, regulated by IGFBPs.
- This is biologically distinct from IGF-1 LR3, which bypasses IGFBPs and produces continuous signaling.
- Pulsatile, IGFBP-regulated signaling is less mechanistically aggressive but not risk-free.
- The epidemiological cancer signal for elevated IGF-1 still applies, just with milder magnitude.
- MK-677 is the exception — its continuous stimulation profile is closer to LR3 than to other secretagogues.
- Tesamorelin has the most clinical data with no increased cancer signal observed.
- Cycle discipline, dose moderation, age-appropriate use, and screening all apply.
- Do not stack with LR3.
- Less risk than LR3, more risk than not intervening.
This concludes Volume 2. Volume 3 picks up with risk stratification, washout strategy, and where the research is heading.
Module 14 — Three Compounds, Three Kinetics: Picking Inside the GH Secretagogue Class
Research and educational purposes only. Not for human consumption.
Why This Module Exists
Module 13 covered the class. This module goes specific. The three GH secretagogues most relevant to the research community discussion — tesamorelin, CJC-1295, and ipamorelin — have meaningfully different kinetic profiles, even though they all activate the same downstream IGF-1 axis. Understanding those differences is how you make compound-specific decisions instead of class-wide ones.
This module also closes the loop on the pulsatile-vs-sustained discussion, because these three compounds span the range from most-pulsatile (ipamorelin) to most-sustained (CJC with DAC).
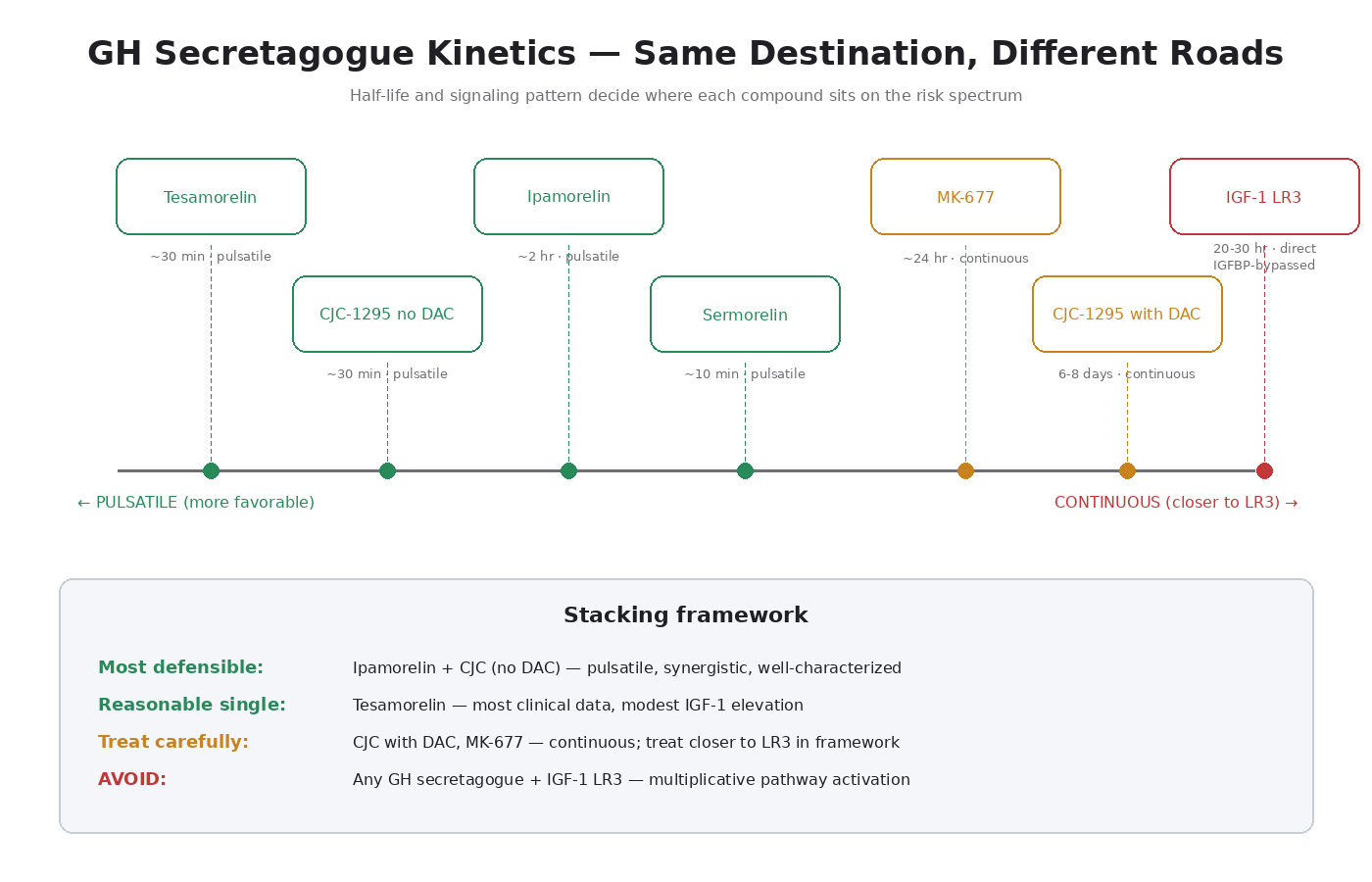
Tesamorelin: The Validated Endpoint
Tesamorelin is a synthetic analog of GHRH (growth hormone-releasing hormone). It has a few amino acid modifications that improve stability and binding affinity, but it functions essentially like enhanced natural GHRH.
Mechanism
- Binds GHRH receptors on pituitary somatotrophs
- Stimulates pulsatile GH release that closely mirrors natural physiology
- Half-life of roughly 30 minutes — enough to enhance natural pulses without producing continuous stimulation
- Subject to all the normal feedback mechanisms (somatostatin, IGF-1 negative feedback)
Clinical Data
Tesamorelin has more clinical trial data than any other compound in this category. It is FDA-approved for HIV-associated lipodystrophy and has been studied in trials lasting 26 to 52 weeks [1].
Key findings from the published trials:
- Modest IGF-1 elevation (~80–100% above baseline, but generally within the physiological range for a healthy adult)
- No increase in cancer incidence over study duration
- Reductions in visceral adipose tissue (the FDA indication)
- Improvements in lipid profile and other metabolic markers
- Generally well-tolerated safety profile
The cancer-related caveats:
- Study durations are months to a year — long-term (5+ years) safety isn’t characterized
- Study populations were specific (HIV-associated lipodystrophy), which may differ from typical research community use
- IGF-1 was kept within physiological range — pushing beyond that produces different risk profiles
- Cancer events in any individual trial cohort are statistically limited, so even modest signals could be missed
Cancer-Pathway Risk Profile
Tesamorelin sits at the more reassuring end of the GH secretagogue spectrum:
- Pulsatile signaling pattern preserved
- Modest IGF-1 elevation
- Established clinical trial data
- All natural feedback mechanisms intact
This doesn’t make it risk-free for chronic recreational use, but the framework treats tesamorelin as one of the better-characterized compounds in the entire research peptide category.
CJC-1295: Two Versions, Very Different Profiles
CJC-1295 is where the nuance gets important. There are two formulations:
CJC-1295 Without DAC (Modified GRF 1-29 / Mod-GRF 1-29)
- Synthetic GHRH analog (the 1–29 fragment with stabilizing modifications)
- Half-life of roughly 30 minutes
- Produces pulsatile GH release similar to natural physiology
- Functionally similar to sermorelin or tesamorelin
This version preserves pulsatility. It enhances natural GH peaks without producing continuous stimulation. From the cancer-pathway perspective, it sits in roughly the same category as tesamorelin — modest, pulsatile IGF-1 elevation through endogenous infrastructure.
CJC-1295 With DAC (Drug Affinity Complex)
- Same GHRH analog with an added maleimidoproprionic acid linker
- The linker binds covalently to serum albumin
- Half-life of roughly 6 to 8 days
- Produces continuous, sustained GH stimulation
- IGF-1 levels rise and stay elevated rather than pulsing
This is a critically different kinetic profile. Continuous GH stimulation produces continuous IGF-1 elevation. The pulsatile-signaling advantage that protects the other compounds in this class is largely lost.
CJC-1295 with DAC sits closer to MK-677 (also continuous) and, in terms of signaling pattern (though still indirect), closer to IGF-1 LR3 than the other GHRH analogs.
The cancer-pathway implication: CJC with DAC and CJC without DAC are not the same compound from a risk-stratification standpoint. Treating them interchangeably misses one of the most important kinetic distinctions in this entire space.
Ipamorelin: The Pulse Enhancer
Ipamorelin is a ghrelin mimetic — it binds the growth hormone secretagogue receptor (GHSR) on pituitary somatotrophs rather than the GHRH receptor. The mechanism is different from CJC, but the output (GH release) is the same [2].
Mechanism
- Binds GHSR (the ghrelin receptor)
- Stimulates GH release that’s complementary to GHRH-driven release
- Half-life of roughly 2 hours
- Highly selective — minimal cortisol, prolactin, or appetite effects (unlike older GHRPs)
- Preserves pulsatile signaling
Why It’s Popular
Ipamorelin is often called the “cleanest” GH secretagogue because of its selectivity. Older ghrelin mimetics (GHRP-2, GHRP-6, hexarelin) also elevated cortisol, prolactin, and appetite in ways that ipamorelin largely avoids.
The Synergy With GHRH Analogs
Ipamorelin is often stacked with CJC-1295 (without DAC) because the two mechanisms synergize. GHRH analogs enhance the “release” signal; ghrelin mimetics enhance the “amplitude” of the release. Together they produce GH pulses that are larger than either alone.
This synergy is mechanistically reasonable and doesn’t change the pulsatile pattern — it just amplifies the existing pulses.
Cancer-Pathway Risk Profile
Ipamorelin sits at the most reassuring end of the GH secretagogue spectrum:
- Highly selective for GH release
- Short half-life preserves pulsatility
- No continuous stimulation
- Minimal off-target hormonal effects
- Stackable with CJC (without DAC) for enhanced natural pulse amplitude
For research community users in the lower-risk profile (younger, no family history, clean screening), ipamorelin + CJC (without DAC) is probably the most defensible GH secretagogue protocol from the cancer-pathway perspective.
The Key Kinetic Comparison
This is the comparison to keep in mind for compound selection. Compound → half-life → signaling pattern → cancer-pathway profile:
- Tesamorelin → ~30 min → pulsatile → most clinical data, modest IGF-1 elevation
- CJC-1295 (no DAC) → ~30 min → pulsatile → similar to tesamorelin
- Ipamorelin → ~2 hours → pulsatile (different receptor) → most selective, minimal off-target effects
- CJC-1295 (with DAC) → 6–8 days → continuous → closer to MK-677 / LR3
- MK-677 → ~24 hours → continuous → closest to LR3 within this class
- IGF-1 LR3 → 20–30 hours → continuous direct receptor → most concerning
The clear pattern: pulsatile compounds with short half-lives sit at the favorable end of the spectrum. Continuous-signaling compounds with long half-lives sit closer to LR3.
Practical Implications For Stacking
A few framework-consistent positions:
Ipamorelin + CJC (no DAC) = the most defensible stack. Both pulsatile, both physiologically reasonable, synergistic mechanism. This is the standard “GH secretagogue cycle” in the research community for good reason.
CJC with DAC = a different conversation. Single weekly injection convenience comes at the cost of pulsatile signaling. If using, treat it more like MK-677 in the risk framework than like the other GHRH analogs.
MK-677 stacked with injectable GH secretagogues = signaling pattern interference. MK-677’s continuous stimulation conflicts with the pulsatile pattern the injectables are trying to create. Probably not a sensible combination.
Any of these stacked with IGF-1 LR3 = the most cancer-pathway-concerning combination. Direct LR3 + endogenous IGF-1 elevation = multiplicative pathway activation. Not recommended.
Tesamorelin alone = the most clinically validated approach. If clinical trial data matters to your risk calculus, tesamorelin is the compound with the most of it.
Cycle Discipline Within This Class
Pulsatile signaling preserves the pulsatile pattern of natural physiology, but chronic uninterrupted use still produces chronic IGF-1 elevation. The framework still applies:
- Time-limited cycles (8–16 weeks)
- Substantial washouts (4–8 weeks between cycles)
- Monitor IGF-1 and IGFBP-3 if doing chronic research use
- Pay attention to absolute IGF-1 levels — staying in the physiological range matters more than the absolute “fast” effect
- Discontinue if any cancer-relevant symptoms or screening findings arise
The Honest Bottom Line
Within the GH secretagogue class, the compounds split into two functional categories:
- Pulsatile-preserving (tesamorelin, CJC no DAC, ipamorelin, sermorelin) — enhance natural GH/IGF-1 pulses, maintain physiological signaling patterns, sit at the more defensible end of the cancer-pathway risk spectrum.
- Continuous-signaling (CJC with DAC, MK-677) — produce sustained GH stimulation, lose the pulsatile-signaling advantage, sit closer to LR3 in terms of mechanistic profile.
The most defensible protocols use pulsatile-preserving compounds in time-limited cycles at moderate doses. The least defensible use continuous-signaling compounds chronically, especially stacked with LR3.
Key Takeaways
- Tesamorelin is the most clinically validated GH secretagogue, with FDA approval and trial data up to 52 weeks.
- CJC-1295 has two versions with critically different kinetics — without DAC (pulsatile, 30 min) and with DAC (continuous, 6–8 days).
- Ipamorelin is highly selective for GH release with minimal off-target hormonal effects.
- Pulsatile-preserving compounds maintain natural physiology and sit at the more defensible end of the spectrum.
- Continuous-signaling compounds (CJC with DAC, MK-677) sit closer to LR3 mechanistically.
- Ipamorelin + CJC (no DAC) is the most defensible stack from a cancer-pathway perspective.
- Do not stack any GH secretagogue with IGF-1 LR3.
- Cycle discipline applies across the entire class.
- Compound-level mechanism specificity, not class-level generalization, is what makes the framework actionable.
Next up: Module 15 — Risk Stratification.
Module 15 — Same Compound, Different You: Why Biological Context Decides Everything
Research and educational purposes only. Not for human consumption.
Why This Module Exists
The first 14 modules covered mechanism. This module covers context. Mechanism doesn’t change based on who’s using the peptide — but the biological context the mechanism is landing in changes everything.
A 25-year-old with no family history and clean screening is in a fundamentally different risk position than a 55-year-old with a first-degree relative who had hormone-sensitive cancer. Same compound, same mechanism, very different risk profile.
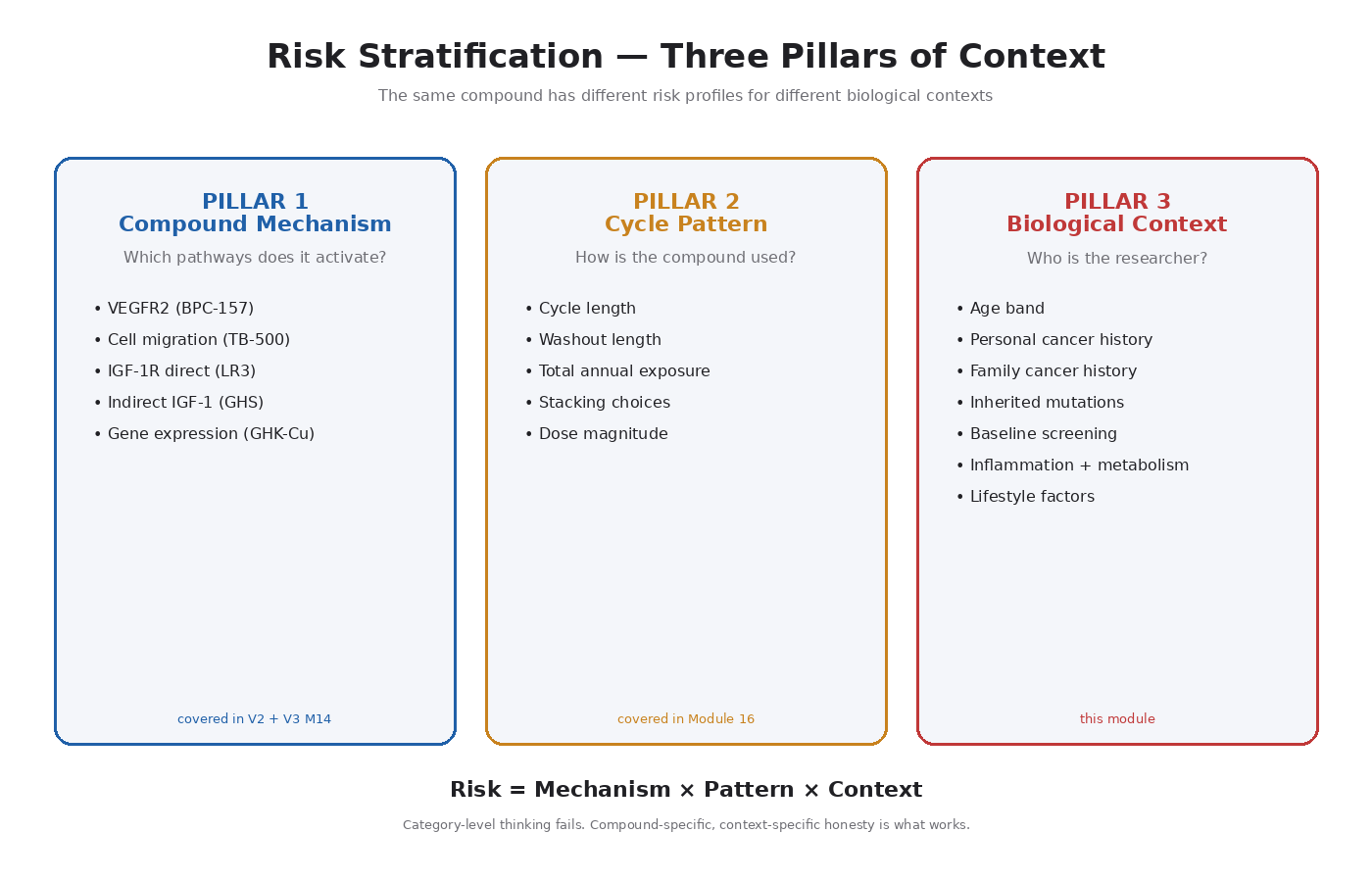
The Three Pillars of Risk Context
The cancer-pathway risk of any growth-signaling peptide depends on three independent factors:
- The mechanism of the compound — covered in V2 Modules 9–13 plus V3 Module 14
- The cycle and dose pattern — covered in Module 16
- The biological context — this module
This module focuses on the third pillar. The key inputs:
- Age
- Personal cancer history
- Family cancer history
- Inherited mutation status
- Baseline screening and surveillance status
- Active inflammatory or metabolic conditions
- Lifestyle factors
Age As a Risk Modifier
Cancer is statistically a disease of cumulative cellular damage. Every cell division creates some risk of replication error. Every environmental exposure adds some DNA damage. Tumor suppressor systems lose efficiency over time.
The consequence: subclinical neoplasms are statistically more common with age [3]. By the time someone reaches their 50s, the probability of having microscopic, undetected neoplastic lesions somewhere in the body is substantially higher than at 25.
Specific examples:
- Colonic adenomas — present in roughly 30 percent of adults over 50
- Prostate microcarcinomas — found in autopsy studies of men over 60 at rates approaching 50 percent (most never become clinically significant)
- Breast atypias and DCIS — increasing prevalence with age
- Thyroid nodules — increasingly common with age
- Skin lesions — accumulating UV damage produces increasing precancerous rates
The framework implication: the cancer-pathway concern of growth-signaling peptides is largely about feeding existing or precancerous lesions. The probability of having such a lesion is age-dependent.
Age-Banded Considerations
Under 30: Low subclinical neoplasm probability, tumor suppressor systems intact, risk dominated by family history and compound selection.
30 to 45: Risk picture shifts, family history weighted more, age-appropriate screening starts to matter, compound selection more important.
45 to 60: Subclinical neoplasm probability rises significantly, baseline screening essential before chronic use, risk-stratification becomes the central framing, IGF-1 LR3 deserves much more careful consideration.
Over 60: Conservative position dominates, comprehensive baseline screening before any chronic use, compound selection narrows substantially, framework leans toward lower-mechanism-impact compounds (GHK-Cu) and away from broad-pathway-activation compounds (LR3).
Family History As a Risk Signal
Family history is one of the most informative inputs to risk-stratification, and it works in two ways:
Statistical signal. A first-degree relative with cancer increases your statistical risk for the same cancer type.
Mechanistic signal. A strong family history often suggests an inherited mutation in a tumor suppressor pathway — even when no specific mutation has been identified. Your biological context may include compromised brakes from the start.
Specific High-Concern Family Patterns
- Two or more first-degree relatives with breast or ovarian cancer — possible BRCA1/2
- Early-onset breast cancer (under 50) in a relative — possible inherited mutation
- Multiple cancers in a single relative (especially TP53-spectrum) — possible Li-Fraumeni syndrome
- Multiple relatives with colorectal cancer, especially young — possible Lynch syndrome
- Pattern of breast + prostate + pancreatic cancer in one family — possible BRCA2
- Family history of breast, prostate, colorectal, or endometrial cancer — IGF-1-coupled cancers [4]
If any of these patterns apply, the conversation isn’t just about “be cautious” — it’s about whether direct IGF-1R activators (LR3 specifically) make sense at all.
Personal Cancer History
If you have a personal history of cancer — currently active, in remission, or treated — the growth-signaling peptide conversation changes fundamentally.
Active cancer or recent treatment: Growth-signaling peptides operate on the same pathways the cancer is using or recovering from. The conservative position is unequivocal — these compounds are not appropriate in this context.
Cancer history with long-term remission: The picture is more nuanced. Time elapsed, cancer type, treatment received, and current surveillance all factor in. This is a conversation that requires oncological input.
Cancer history of IGF-1-coupled types: Breast, prostate, colorectal, and endometrial cancers all have established IGF-1R involvement. History of any of these substantially shifts the risk calculus around IGF-1-activating peptides.
Inherited Mutations And Predisposition Syndromes
If genetic testing has identified an inherited mutation, that information is the most direct input to risk-stratification:
- BRCA1 or BRCA2 carriers — DNA repair compromised
- Lynch syndrome (MMR mutations) — mismatch repair compromised
- Li-Fraumeni syndrome (TP53 mutation) — central tumor suppressor already compromised
- Cowden syndrome (PTEN mutation) — main brake on PI3K/Akt/mTOR compromised; most directly relevant for peptide users
Baseline Screening Before Chronic Use
Chronic use of any growth-signaling peptide should be preceded by appropriate baseline screening. Not because peptides cause cancer, but because the cancer-pathway concern is about feeding existing or precancerous lesions.
Age-Appropriate Screenings
- Colonoscopy — starting at age 45 per current guidelines
- Mammography (for women) — age-appropriate baseline
- Prostate exam and PSA (for men over 50)
- Pap and HPV testing (for women)
- Skin exam — full-body dermatologic exam
- Thyroid palpation and TSH
Bloodwork Worth Having
- Baseline IGF-1 and IGFBP-3 — establishes starting point before any GH/IGF-1 intervention
- Complete blood count (CBC)
- Comprehensive metabolic panel (CMP)
- Fasting insulin and glucose
- Inflammatory markers (CRP, ESR) [5]
- Thyroid panel — TSH, free T4, free T3
Symptoms To Monitor During Use
- Unexplained weight loss
- New or changing lumps or nodules
- Persistent changes in bowel habits
- Persistent cough, hoarseness, or difficulty swallowing
- Unexplained bleeding
- New persistent pain
- Persistent fatigue out of proportion to context
- Changes in skin lesions
The Inflammation And Metabolic Context
Chronic inflammation is itself a cancer risk factor. Chronic NF-κB activation drives both inflammation and tumor-promoting signaling. Conditions that produce chronic inflammation elevate baseline cancer risk.
Metabolic dysfunction — insulin resistance, hyperinsulinemia, type 2 diabetes — produces chronic hyperinsulinemia that activates IGF-1R (overlapping receptors). This raises baseline cancer risk independently of any peptide use.
The framework implication: improving metabolic health and reducing chronic inflammation is part of responsible research peptide use.
Lifestyle Factors That Affect Baseline Risk
- Alcohol consumption — increases risk for multiple cancer types
- Tobacco use — clearly increases multiple cancer risks
- UV exposure — skin cancer risk
- Obesity — drives metabolic dysfunction, inflammation, hormonal changes
- Sedentary behavior — independent risk factor
- Diet quality — multiple complex relationships
- Sleep quality — circadian disruption is associated with cancer risk
- Hormonal exposure history — estrogen exposure, anabolic steroid use, etc.
Lifestyle factors set the baseline. Peptides operate on that baseline.
A Personal Risk Profile Worksheet
Walking through these questions honestly is the practical application of this module:
- What is my age range? (Under 30 / 30–45 / 45–60 / over 60)
- Do I have any personal cancer history? (None / treated / active)
- What is my family cancer history? (None / single distant / one first-degree / multiple / known syndrome)
- Has genetic testing identified any inherited mutations? (No / yes — specify)
- What is my baseline screening status? (Up to date / partial / none)
- Do I have known inflammatory or metabolic conditions?
- What is my baseline IGF-1?
- What is my lifestyle baseline? (Healthy / mixed / multiple risk factors)
Key Takeaways
- Cancer-pathway risk has three pillars: compound mechanism, cycle pattern, biological context.
- Age increases the probability of subclinical neoplasms.
- Family history works as both a statistical signal and a mechanistic signal.
- Personal cancer history fundamentally shifts the risk conversation.
- Inherited mutations (BRCA1/2, Lynch, Li-Fraumeni, Cowden) are the strongest single inputs.
- Baseline screening should precede chronic use.
- IGF-1 / IGFBP-3 bloodwork establishes the starting point.
- Chronic inflammation and metabolic dysfunction elevate baseline cancer risk.
- Lifestyle factors set the baseline; peptides operate on top of it.
- Some compounds may not be appropriate for some risk profiles.
Next up: Module 16 — Washout Strategy.
Module 16 — The Single Most Important Practice: Why Cycling Isn’t Optional
Research and educational purposes only. Not for human consumption.
Why This Module Exists
Chronic, uninterrupted growth signaling is the most concerning configuration for cancer-pathway risk. The single most important countermeasure available to a researcher is the same one nature uses: interruption. Natural physiology is pulsatile. Hormones rise and fall. Receptors get internalized and recycled. Feedback loops kick in and shut down chronically activated pathways. Continuous signaling without recovery is not a state the body is designed to maintain.
Washouts aren’t a “nice to have.” They’re the central risk-reduction practice across the entire growth-signaling peptide category.
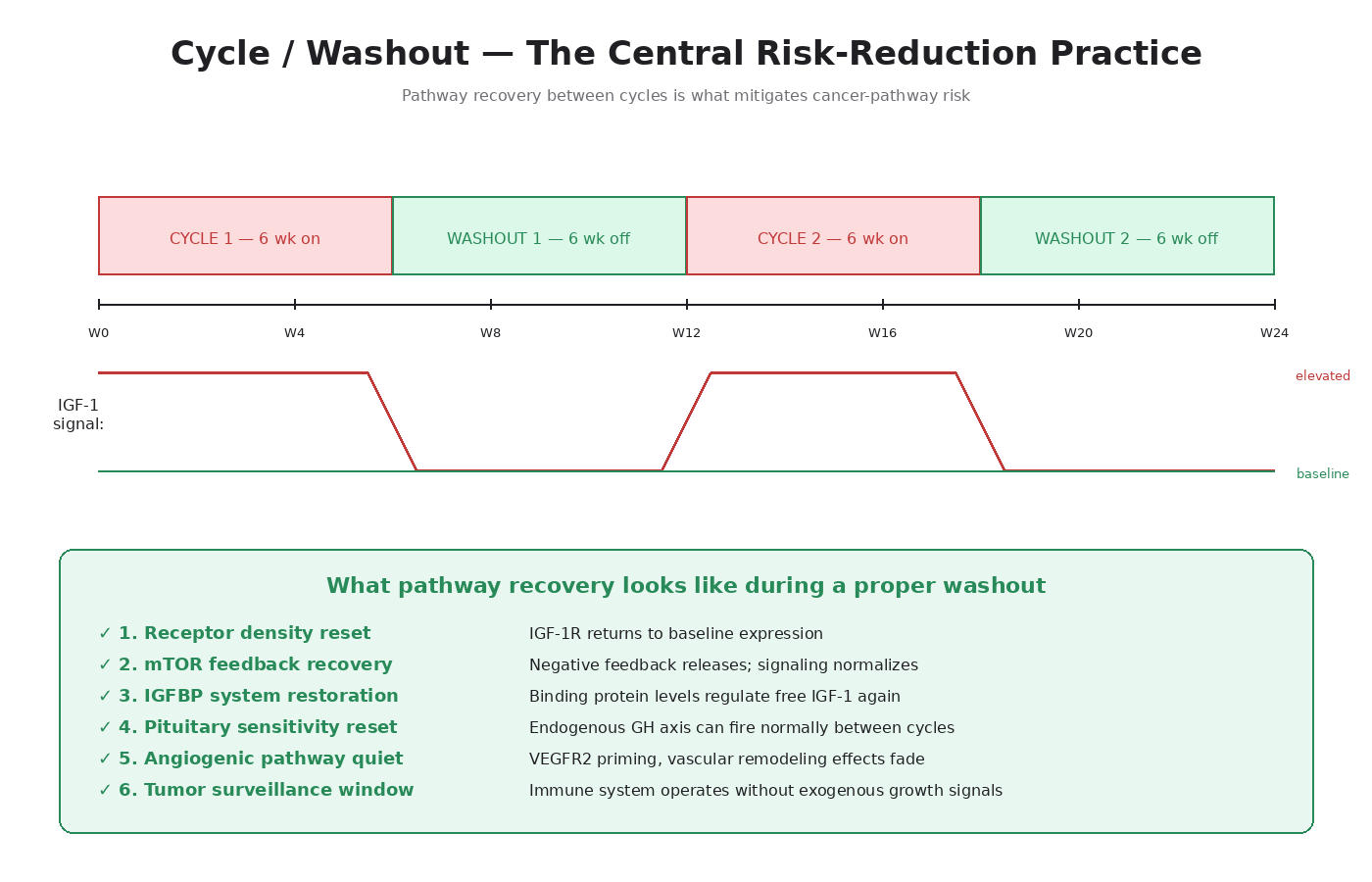
What a Washout Actually Is
A washout is a period of complete non-use following a peptide cycle. The compound clears the body. Receptor densities reset. Downstream signaling returns to baseline. Feedback systems re-establish normal regulation.
A washout is not a “deload.” It’s complete non-use. Receptors don’t reset under continued low-level stimulation — they reset under absence of stimulation.
Rotating between different peptides that hit the same pathway is not a washout. If you cycle off IGF-1 LR3 and switch to GH secretagogues that elevate endogenous IGF-1, you haven’t washed out IGF-1R signaling — you’ve just switched the upstream input. The downstream pathway sees continuous activation.
True washout requires interrupting the pathway, not just the compound.
Why Pathway Recovery Matters Mechanistically
1. Receptor Density Reset
When IGF-1R is chronically activated, the cell downregulates receptor expression to dampen the signal. The downregulation isn’t immediate — it takes weeks to fully manifest — and reversal also takes weeks once stimulation stops. A 2-week cycle followed by a 2-week washout doesn’t allow full receptor recovery. A 6-week cycle followed by a 6-week washout begins to.
2. mTOR Feedback Inhibition Recovery
Chronic mTOR activation triggers a negative feedback loop that suppresses IGF-1R signaling. When the stimulation stops, this feedback releases — but the IGF-1R signaling often rebounds above baseline before normalizing.
3. IGFBP Restoration
In normal physiology, IGFBPs sequester most circulating IGF-1. Chronic GH/IGF-1 elevation alters IGFBP levels and ratios. A washout allows the IGFBP system to re-establish its normal regulatory role.
4. Hypothalamic-Pituitary Sensitivity Recovery
For GH secretagogue users, chronic exogenous stimulation can desensitize the pituitary’s response to natural GHRH. Washouts allow that sensitivity to recover.
5. Angiogenic Pathway Normalization
For BPC-157 and TB-500 users, the VEGFR2 upregulation and angiogenic priming don’t immediately reverse when the peptide is discontinued. Vascular remodeling effects persist for weeks.
6. Tumor Surveillance Window
The immune system’s tumor surveillance operates better when growth-signaling pathways are at baseline. A washout provides a window where any early neoplastic activity is not being fed by exogenous growth signals.
Cycle-to-Washout Ratios
The general framework principle: the washout should be at least as long as the cycle. A 4-week cycle warrants at least a 4-week washout. An 8-week cycle warrants at least 8 weeks off.
For higher-risk compounds (IGF-1 LR3 specifically), the conservative position is washouts longer than the cycle.
Compound-Specific Cycle Guidance
BPC-157 / TB-500: 4–6 weeks on, 4–6 weeks off. For acute injury indications, the cycle can be limited to the healing window.
IGF-1 LR3: 4 weeks on max, 6–8 weeks washout, with total annual exposure kept limited. Some researchers run shorter cycles (2–3 weeks) with proportional washouts.
GH secretagogues (pulsatile — tesamorelin, CJC no DAC, ipamorelin): 8–16 week cycles with 4–8 weeks off.
CJC-1295 with DAC / MK-677: Treat closer to LR3 in framework — shorter cycles, longer washouts.
GHK-Cu: The cancer-pathway risk profile is fundamentally different. General cycling principles still apply, but the cancer-pathway driver is less acute.
The “Bridging” Problem
A practice that comes up in the community: using one peptide to “bridge” between cycles of another, so the user is never fully off all compounds.
The framework position: bridging defeats the purpose of washouts. If the goal is pathway recovery, the pathway needs to be quiet. Bridging maintains some level of growth-signaling activation continuously, even when the specific compound changes.
The defensible alternative to bridging: alternate cycles of compounds that hit different pathways. A cycle of growth-signaling peptide followed by a cycle of GHK-Cu followed by another growth-signaling cycle gives the growth pathway full rest while still allowing other research applications.
Markers To Track Across Cycles
Bloodwork before each cycle:
- IGF-1 and IGFBP-3 (for any GH/IGF-1-modulating cycle)
- Fasting insulin and glucose
- Complete blood count
- Comprehensive metabolic panel
- Inflammatory markers (CRP)
Comparison points:
- Does IGF-1 return to baseline during washouts?
- Is the trajectory of IGF-1 across multiple cycles stable, or drifting upward?
- Are inflammatory markers staying in check?
- Is metabolic health stable?
If IGF-1 doesn’t return to baseline between cycles, the washouts are too short.
The Total Annual Exposure Concept
How many weeks per year is the researcher administering growth-signaling peptides? How many weeks per year is the body in full pathway-recovery mode?
For a low-risk-profile researcher using moderate-mechanism compounds, total annual exposure of perhaps 16–24 weeks (with 28–36 weeks off) is a defensible framework. For higher-risk profiles or higher-mechanism compounds, annual exposure of 8–16 weeks with longer rest periods is more conservative.
When To Extend a Washout
- IGF-1 hasn’t returned to baseline
- Inflammatory markers are elevated
- Any new symptoms warranting investigation
- Any new findings on screening
- Significant life stressors that compromise immune function or sleep
- Onset of any condition that might affect baseline cancer risk
When To Discontinue a Compound
- Any cancer-suggestive symptoms appearing during use
- Any new findings on screening
- IGF-1 trending upward across multiple cycles despite stable doses
- Family discovery of a relevant inherited mutation
- Change in personal risk profile
Discontinuation is a defensible research decision, not a failure mode.
Key Takeaways
- Chronic, uninterrupted growth signaling is the most concerning configuration.
- Washouts are complete non-use periods that allow receptor reset, feedback recovery, and pathway normalization.
- Rotating compounds that hit the same pathway is not a washout — pathway-level rest is what matters.
- Standard framework: washout at least as long as the cycle; for higher-risk compounds, longer.
- Bridging defeats the purpose.
- Track IGF-1 and other markers across cycles.
- Total annual exposure matters as much as individual cycle structure.
- Some signals warrant extending a washout; some warrant discontinuing a compound entirely.
- Cycle discipline is the single most important risk-reduction practice in this category.
Next up: Module 17 — Peptides as Cancer Adjuncts.
Module 17 — The Other Side: Peptides Already Being Studied to Help Treat Cancer
Research and educational purposes only. Not for human consumption.
Why This Module Exists
This course has spent sixteen modules on the cancer-pathway concerns associated with growth-signaling peptides. It would be incomplete without acknowledging the other side: there is a substantial and growing body of research on peptides being investigated as adjuncts to cancer treatment, as supportive care during chemotherapy and radiation, and as research tools for understanding cancer biology.
The framework has always been mechanism-specific, not category-wide. Some peptides operate on cancer-promoting pathways. Others operate on tumor-suppressing pathways, immune-enhancing pathways, or pathways that mitigate the collateral damage of conventional treatment.
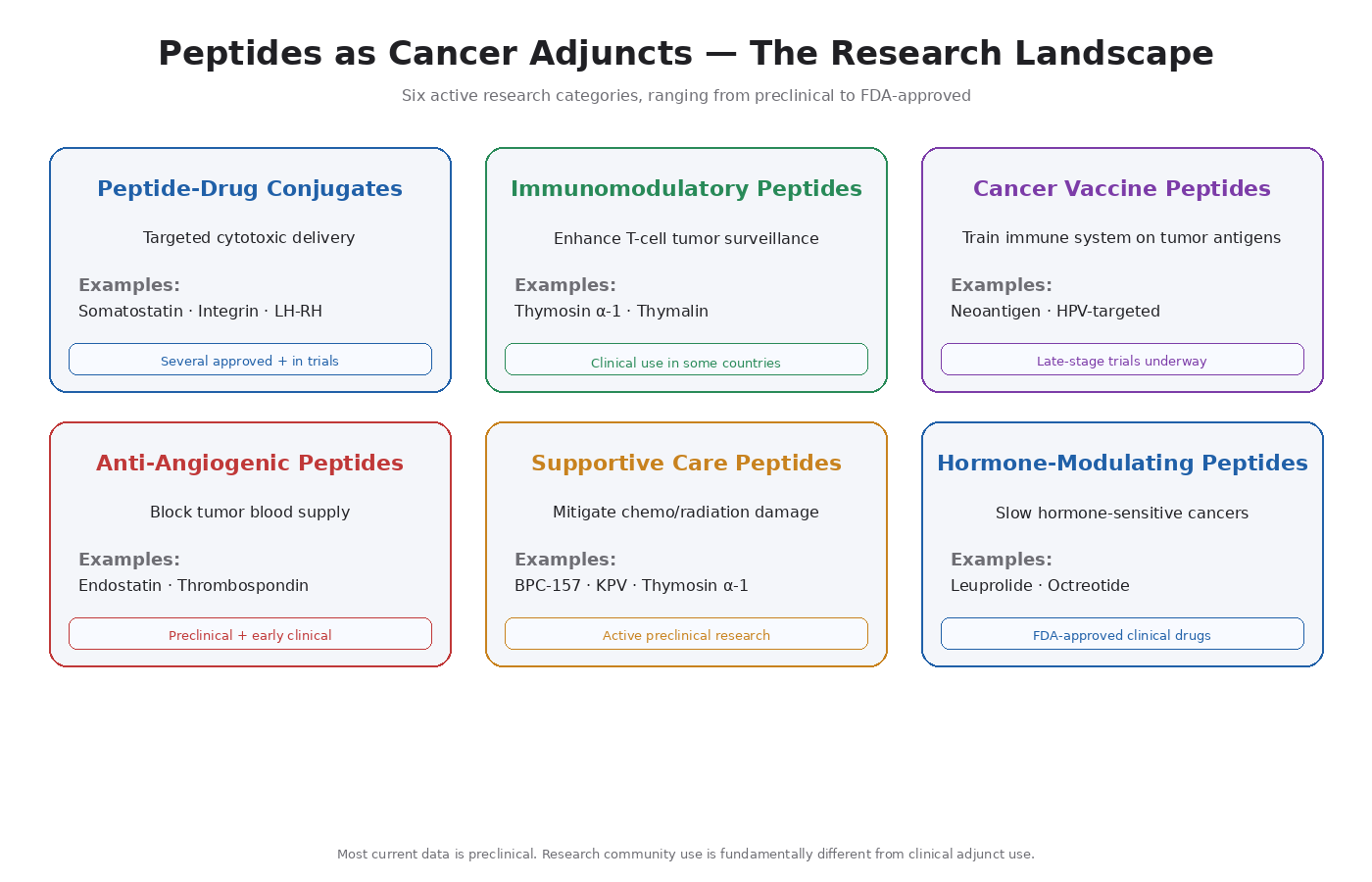
The Categories of Peptide Cancer Research
1. Peptide-Drug Conjugates (PDCs)
Targeted delivery of cytotoxic drugs using peptides as homing molecules. The peptide recognizes a receptor overexpressed on tumor cells; the attached cytotoxic payload kills the cell once delivered.
Examples in development:
- Somatostatin analog conjugates for neuroendocrine tumors
- Integrin-targeting peptide conjugates for various solid tumors
- LH-RH analog conjugates for hormone-receptor-positive cancers
2. Immunomodulatory Peptides As Adjuncts
- Thymosin alpha-1 — enhances T-cell function, used clinically in some countries for cancer adjunct therapy [6]
- Thymalin — similar T-cell enhancement profile
- Various thymic peptides — broader category, multiple compounds in research
3. Cancer Vaccine Peptides
Short peptide sequences from tumor antigens used to train the immune system to recognize cancer cells [7].
- Personalized neoantigen vaccines (peptides specific to mutations in an individual’s tumor)
- Shared tumor antigen vaccines (peptides targeting commonly mutated proteins)
- HPV-targeted vaccines for HPV-driven cancers
4. Anti-Angiogenic Peptides
Peptides that block angiogenesis — the opposite mechanism of BPC-157 and TB-500.
- Endostatin-derived peptides
- Thrombospondin-derived peptides
- VEGFR-blocking peptide fragments
5. Supportive Care Peptides
Peptides that mitigate the side effects of chemotherapy and radiation.
- BPC-157 — research into mitigating chemotherapy-induced gastrointestinal damage
- Thymosin alpha-1 — supporting immune function during chemotherapy
- KPV — anti-inflammatory effects in radiation-induced tissue damage
6. Hormone-Modulating Peptides
- LH-RH agonists/antagonists (leuprolide, degarelix) — used clinically in prostate cancer and breast cancer
- Somatostatin analogs (octreotide, lanreotide) — used clinically in neuroendocrine tumors
Specific Peptides the Research Community Discusses
BPC-157 As a Chemotherapy Adjunct
The C26 colon adenocarcinoma cachexia work is the most-cited evidence [8]. Researchers administering BPC-157 to mice with C26 tumors observed:
- Counteracted muscle wasting (cachexia)
- Preserved myogenesis
- Prolonged survival
- Reduced pro-cachectic cytokine elevations (IL-6, TNF-alpha)
- Did not accelerate tumor growth in that specific model
Honest framing: preclinical, mostly animal model data. Mechanistically interesting and consistent. Doesn’t mean BPC-157 treats cancer in humans.
Thymosin Alpha-1 In Cancer Treatment
Thymosin alpha-1 has been used clinically in some countries (approved in Italy and several others, though not in the US) for various indications including supporting cancer treatment. Research base includes:
- Enhanced T-cell function
- Modulation of innate and adaptive immunity
- Some evidence of synergy with chemotherapy in specific contexts
- Studies in hepatocellular carcinoma, melanoma, and others
KPV And Inflammation-Related Cancer Pathways
KPV has anti-inflammatory effects through NF-κB modulation. Since chronic inflammation is a major cancer driver, there’s research interest in KPV and similar anti-inflammatory peptides for cancer prevention and adjunct contexts. Most of this work is preclinical.
GHK-Cu Research Beyond Cosmetics
The Pickart gene expression work [9] showed striking tumor suppressor upregulation and cancer enhancer downregulation in cell line studies. Clinical translation hasn’t yet happened, but the mechanism is interesting.
The Research Frontier — What’s Coming
- CAR-T Adjacent Peptide Approaches — peptides that enhance or modulate engineered T-cell therapies
- Tumor Microenvironment Modulation — peptides that make tumors more visible to the immune system
- Senolytic And Senomorphic Peptides — selectively eliminate or modify senescent cells
- Cancer-Associated Fibroblast Modulators — non-cancer cells in the tumor microenvironment that support cancer growth
- Personalized Neoantigen Vaccines — likely to reach clinical relevance in the next several years
What This Means for the Research Community
The “all peptides are dangerous in cancer contexts” framing is wrong. Peptide-based cancer interventions are an active and growing area of research. Some peptides may eventually become cancer drugs in their own right.
The “research community peptide use is the same as clinical cancer adjunct use” framing is also wrong. Clinical research uses specific compounds, specific doses, specific durations, specific patient populations, with active medical monitoring. Recreational research use is a very different context.
The framework’s mechanism-specificity holds up. GHK-Cu being studied as a possible cancer-protective intervention isn’t a contradiction of IGF-1 LR3 being concerning for cancer-pathway risk. They’re different compounds operating on different pathways.
The honest middle position is durable. Peptides don’t cause cancer. Some peptides feed cancer pathways. Some peptides modulate pathways in ways that may eventually have anti-cancer applications.
Caveats On The Research Frontier
- Preclinical doesn’t equal clinical. Most data on research-community peptides as cancer adjuncts is preclinical.
- Adjunct use isn’t the same as standalone treatment. No peptide in the research community is a cancer treatment in any clinical sense.
- Regulatory and safety pathways matter. Translating clinical cancer research to off-label research use in healthy researchers requires substantial caution.
- Hype vs. signal is a real problem. Weight peer-reviewed published data above community claims, and clinical trial data above preclinical mechanism papers.
Where To Watch Going Forward
- PubMed — peer-reviewed published research
- ClinicalTrials.gov — registry of clinical trials
- Conference proceedings — AACR, ASCO, ESMO
- Peer-reviewed journals — Cancer Research, Clinical Cancer Research, Journal of Peptide Science
Key Takeaways
- Peptide-based cancer interventions are an active and growing area of clinical and preclinical research.
- Categories include peptide-drug conjugates, immunomodulators, cancer vaccines, anti-angiogenic peptides, supportive care peptides, and hormone-modulating peptides.
- Several compounds the research community discusses (BPC-157, thymosin alpha-1, KPV, GHK-Cu) have research interest as cancer adjuncts.
- Most current data is preclinical — clinical translation isn’t guaranteed.
- The framework’s mechanism-specificity holds: some peptides feed cancer pathways, others modulate pathways with possible anti-cancer applications, both can be true.
- Honest framing requires both cancer-pathway caution and cancer-adjunct curiosity simultaneously.
Closing Note on the Whole Course
You’ve made it through seventeen modules covering the molecular biology of cancer, the specific pathways tumors hijack, the mechanisms of the major research community peptides, the framework for risk stratification, and the cycling discipline that actually mitigates risk.
The single most important thing to take away from all of this: mechanism matters, context matters, and the framework only works if you actually apply it to yourself.
Peptides do not cause cancer. Specific peptides operate on specific pathways with specific cancer-pathway overlaps. The risk profile of any compound depends on the user’s biological context, cycling discipline, and total exposure. Category-level thinking in either direction fails. Mechanism-specific, context-specific honesty is what works.
The research community is at its best when it operates from this level of understanding. The community is at its worst when it operates from rumor, fear, or unconditional cheerleading. This course was built to support the first mode and counter the second.
Stay curious. Stay disciplined. Stay honest about your own biological context. And let the research evolve your framework as new data emerges.
Research and educational purposes only. Not for human consumption.
Closing Note on the Whole Course
You’ve made it through seventeen modules covering the molecular biology of cancer, the specific pathways tumors hijack, the mechanisms of the major research community peptides, the framework for risk stratification, and the cycling discipline that actually mitigates risk.
The single most important thing to take away from all of this: mechanism matters, context matters, and the framework only works if you actually apply it to yourself.
Peptides do not cause cancer. Specific peptides operate on specific pathways with specific cancer-pathway overlaps. The risk profile of any compound depends on the user’s biological context, cycling discipline, and total exposure. Category-level thinking in either direction fails. Mechanism-specific, context-specific honesty is what works.
The research community is at its best when it operates from this level of understanding. The community is at its worst when it operates from rumor, fear, or unconditional cheerleading. This course was built to support the first mode and counter the second.
Stay curious. Stay disciplined. Stay honest about your own biological context. And let the research evolve your framework as new data emerges.
Research and educational purposes only. Not for human consumption.
References
- Falutz J, Allas S, Blot K, et al. Metabolic effects of a growth hormone-releasing factor in patients with HIV. N Engl J Med. 2007;357(23):2359–2370. PMID: 18057338.
- Raun K, Hansen BS, Johansen NL, et al. Ipamorelin, the first selective growth hormone secretagogue. Eur J Endocrinol. 1998;139(5):552–561. PMID: 9849822.
- Welch HG, Black WC. Overdiagnosis in cancer. J Natl Cancer Inst. 2010;102(9):605–613. PMID: 20413742.
- Pollak MN. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8(12):915–928. PMID: 19029956.
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–867. PMID: 12490959.
- Garaci E, Pica F, Rasi G, Favalli C. Thymosin alpha 1 in the treatment of cancer: from basic research to clinical application. Int J Immunopharmacol. 2000;22(12):1067–1076. PMID: 11150840.
- Hu Z, Ott PA, Wu CJ. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol. 2018;18(3):168–182. PMID: 29226910.
- Kang EA, Han YM, An JM, et al. BPC157 as potential agent rescuing from cancer cachexia. Curr Pharm Des. 2018;24(18):1947–1956. PMID: 29512451.
- Pickart L, Margolina A. Regenerative and protective actions of the GHK-Cu peptide in the light of the new gene data. Int J Mol Sci. 2018;19(7):1987. PMID: 29986520.
Other parts in this series: