Part 2 of the three-part research-use series. Part 1 covered cancer biology foundations and the major pathways tumors hijack. This part finishes the foundations with metastasis and tumor suppressors, then walks through the first four peptide deep dives: BPC-157, TB-500, IGF-1 LR3, and GHK-Cu.
The framework from Part 1 — peptides don't cause cancer, but specific peptides operate on specific cancer-relevant pathways — is applied here compound by compound. By the end of this part you can read any peptide study and place the mechanism inside the framework.
Module 7 — The Migration Switch: How Cancer Learns to Travel
Research and educational purposes only. Not for human consumption.
Why This Module Matters
Roughly 90 percent of cancer deaths are not caused by the primary tumor. They’re caused by metastases. A tumor sitting in one organ can usually be managed surgically. A tumor that has spread to liver, bone, and brain cannot. Whatever lets cancer cells leave their original location and colonize other tissues is, in practical terms, the most lethal ability they acquire.
That ability is mediated by a process called epithelial-mesenchymal transition (EMT) [16]. It’s the molecular machinery that lets a static, settled cell become a migratory one. And it’s the most direct overlap point for TB-500 — thymosin beta-4, the actin-sequestering peptide whose entire function is to promote cell migration.
This module is the one TB-500 users specifically need to read.
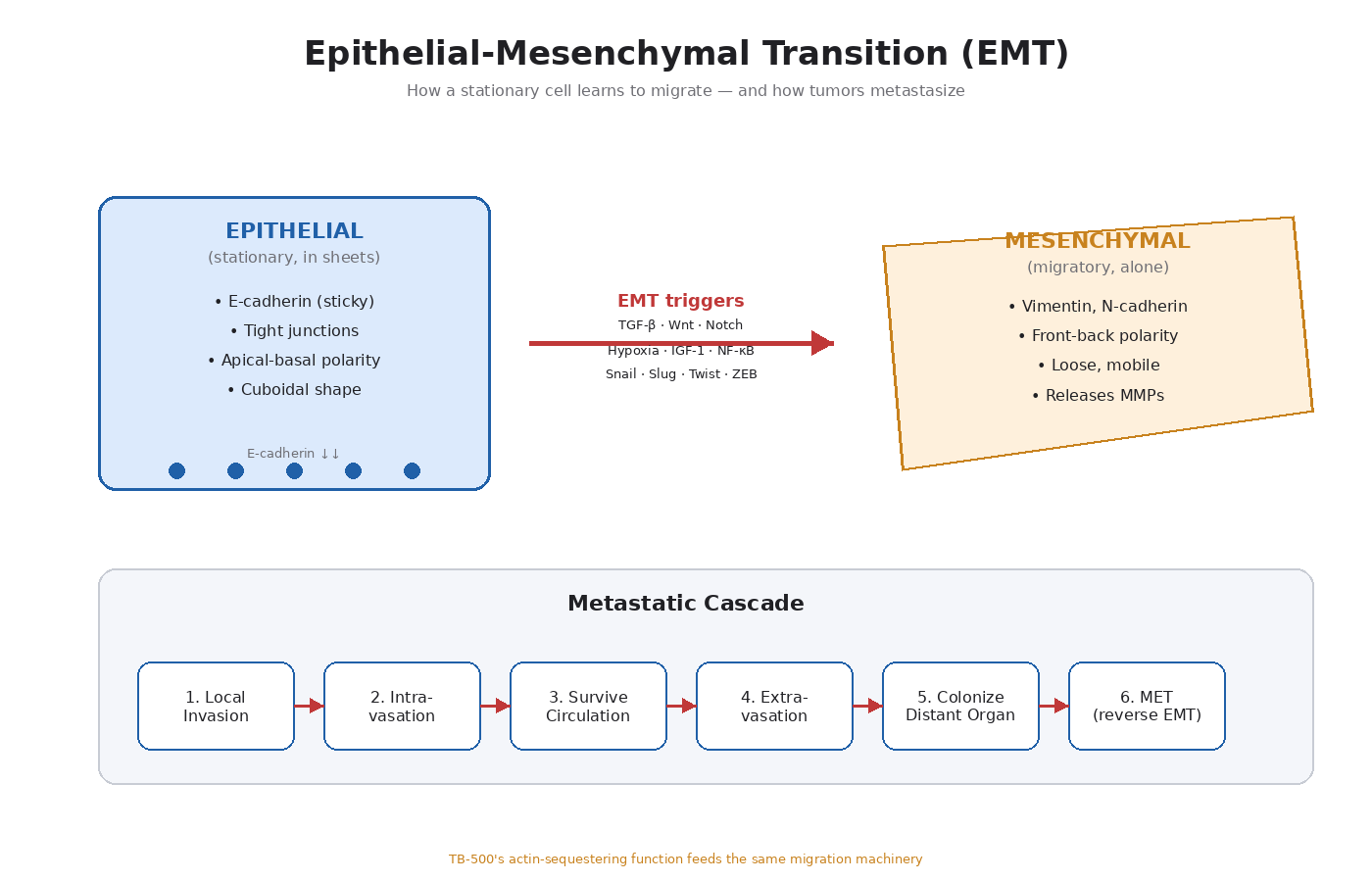
What EMT Actually Is
Your body has two basic types of tissue architecture: epithelial and mesenchymal.
Epithelial cells form sheets and layers. They’re stationary. They stick tightly to each other through specialized cell-cell junctions (tight junctions, adherens junctions, desmosomes). They have apical-basal polarity — a defined “top” and “bottom.” They express E-cadherin, the main adhesion molecule that holds epithelial sheets together. Examples: the lining of your gut, the layer covering your skin, the cells of a breast duct.
Mesenchymal cells are migratory. They don’t form sheets. They don’t stick tightly to each other. They have front-back polarity oriented toward direction of movement. They express vimentin (an intermediate filament protein) and N-cadherin instead of E-cadherin. Examples: fibroblasts, immune cells in transit, stem cells migrating during development.
Epithelial-mesenchymal transition (EMT) is the molecular program that lets an epithelial cell switch into mesenchymal mode. It happens naturally during embryo development (when cells need to migrate to form organs) and during wound healing (when epithelial cells at the edge of a wound need to crawl into the gap).
It also happens in cancer. And when it does, it’s the trigger for metastasis.
The Hallmarks of EMT, Molecularly
When a cell undergoes EMT, several things happen in coordinated sequence:
- Loss of E-cadherin — the cell stops sticking to its neighbors.
- Loss of tight junctions and desmosomes — the cell loses its sheet architecture.
- Loss of apical-basal polarity — the cell can move in any direction.
- Cytoskeletal rearrangement — actin filaments reorganize to drive migration.
- Upregulation of N-cadherin and vimentin — mesenchymal markers come online.
- Secretion of matrix metalloproteinases (MMPs) — enzymes that degrade the surrounding extracellular matrix.
- Activation of EMT-inducing transcription factors — Snail, Slug, Twist, ZEB1, ZEB2 — which orchestrate the whole program.
The transcription factors are the master switches. Snail and Slug, in particular, are the proteins that directly repress E-cadherin expression. When Snail or Slug get activated, E-cadherin gets shut off, and the cell starts losing its epithelial identity.
What Triggers EMT in Cancer
Tumor cells use the same triggers that embryonic cells and wound-healing cells use. The main inducers:
- TGF-β (transforming growth factor beta) — the most studied EMT inducer
- Wnt signaling — drives both EMT and stemness
- Notch signaling — particularly important in breast cancer EMT
- Hypoxia (low oxygen) — HIF-1alpha induces Snail and other EMT factors
- Growth factors that activate MAPK/ERK and PI3K/Akt — including IGF-1, EGF, FGF, HGF
- Inflammatory signals — TNF-alpha, IL-6, NF-κB activation
- Mechanical signals — stiffness of the surrounding extracellular matrix
Notice how many of these overlap with pathways we’ve already covered. PI3K/Akt activation (Module 4) drives EMT. MAPK/ERK activation (Module 5) drives EMT. IGF-1R signaling (Module 6) drives EMT. Any peptide that activates these pathways is, by extension, contributing to the upstream machinery of EMT.
The Metastatic Cascade, Step by Step
Once a tumor cell has undergone EMT, it can leave the primary tumor. The full metastatic cascade looks like this:
- Local invasion — the cell breaks through the basement membrane (the layer that normally separates epithelial tissue from underlying tissue).
- Intravasation — the cell enters a blood vessel or lymphatic vessel.
- Survival in circulation — the cell has to survive shear stress and immune surveillance in the bloodstream.
- Extravasation — the cell exits the vessel at a distant site.
- Colonization — the cell adapts to the new microenvironment and starts dividing.
- Mesenchymal-epithelial transition (MET) — the cell often reverses EMT to re-establish epithelial growth at the new site.
The fact that the cell can reverse EMT (called MET) at the destination is important. Cancer cells aren’t permanently committed to mesenchymal mode. They use it as a transient tool for migration, then switch back when they need to colonize. This plasticity is one of the most difficult features of metastatic cancer to target therapeutically.
TB-500 (Thymosin Beta-4) and EMT
TB-500 is the synthetic fragment of thymosin beta-4 (TB4), a 43-amino-acid peptide present in essentially every cell in the human body. Its main functions:
- Actin sequestration — TB4 binds G-actin (monomeric actin) and prevents it from polymerizing into F-actin (filaments) prematurely. This regulates cytoskeletal dynamics.
- Cell migration — by controlling actin polymerization, TB4 lets cells migrate efficiently.
- Angiogenesis — drives endothelial cell migration and VEGF expression (Module 3).
- Anti-inflammatory effects — modulates NF-κB and inflammatory cytokine production.
- Tissue regeneration — supports wound healing across multiple organ systems.
Here’s where it gets directly relevant to this module. The oncology literature on endogenous TB4 is substantial:
- TB4 is overexpressed in multiple cancers, including melanoma, colorectal cancer, non-small-cell lung cancer, hepatocellular carcinoma, oral squamous cell carcinoma, renal cell carcinoma, and others [17].
- In hepatoblastoma research, TB4 has been shown to directly drive EMT through downregulation of E-cadherin and beta-catenin.
- In oral squamous cell carcinoma, TB4 induces proliferation, invasion, and full EMT.
- TB4 expression correlates with tumor aggressiveness and metastatic potential in multiple cancer types.
- It activates the integrin-linked kinase (ILK) pathway, which is itself an EMT driver.
The most important nuance: TB4 appears to be a marker of aggressive tumors more than a transformative agent. Cancer cells upregulate it because they need its actin-sequestering, migration-promoting, angiogenic properties. There is no published evidence that exogenous TB-500 transforms a healthy cell into a cancer cell.
But the pathway overlap is direct. TB4 is one of the proteins tumors use to metastasize. Administering more of it to a body with undetected cancer is the most mechanistically concerning peptide-cancer interaction in this entire course, alongside IGF-1 LR3.
Why the TB-500 Conversation Is Different from the BPC-157 Conversation
Both peptides drive angiogenesis. Both touch growth signaling pathways. So why does TB-500 deserve more caution?
BPC-157’s primary action is upregulating receptor density and signaling sensitivity — it makes existing growth-signal infrastructure work better. It also has documented anti-tumor effects in some specific cancer models. The melanoma cell line data showing VEGF inhibition is real.
TB-500’s primary action is enabling cell migration — and migration is the central process of metastasis. There is no analog to the BPC-157 anti-tumor cell-line data for TB-500. Every cancer study on endogenous TB4 shows it correlates with worse outcomes, more aggressive disease, and higher metastatic potential.
The mechanism of TB-500 is, in oncological terms, almost identical to one of the things tumor cells need to do to spread.
This doesn’t mean TB-500 causes cancer. It means that in the presence of an existing cancer, broad amplification of cell migration machinery is a poor configuration.
Other Peptides That Touch EMT
EMT is downstream of so many signaling pathways that nearly any growth-signaling peptide contributes to it indirectly:
- IGF-1 LR3 drives EMT through PI3K/Akt activation.
- Growth hormone secretagogues indirectly through elevated IGF-1.
- EGF analogs through MAPK/ERK.
- TGF-β-related compounds directly through the most studied EMT pathway.
- BPC-157 has weaker EMT-promoting effects compared to TB-500, though some indirect signaling overlap exists.
Conversely, some peptides actually downregulate EMT pathways:
- GHK-Cu upregulates E-cadherin and downregulates several EMT-related genes in some cancer cell lines [13].
- KPV has anti-inflammatory effects that reduce EMT-promoting cytokines.
- Thymosin alpha-1 modulates immune surveillance, which targets cells undergoing EMT.
Key Takeaways
- Metastasis causes ~90 percent of cancer deaths.
- EMT is the molecular program that lets a settled epithelial cell become a migratory mesenchymal one.
- Loss of E-cadherin is the central event — driven by transcription factors Snail, Slug, Twist, ZEB1/2.
- Triggers include TGF-β, Wnt, Notch, hypoxia, growth factors (IGF-1, EGF, FGF, HGF), inflammation.
- The full metastatic cascade involves local invasion, intravasation, circulation survival, extravasation, colonization, and MET at the destination.
- TB-500 (synthetic thymosin beta-4) is the peptide with the most direct EMT-machinery overlap.
- Endogenous TB4 is overexpressed in melanoma, colorectal, lung, liver, and many other cancers.
- TB4 appears to be a marker and driver of aggressive cancer behavior, not a transformative agent.
- IGF-1 LR3 and growth hormone secretagogues drive EMT indirectly through PI3K/Akt and MAPK/ERK activation.
- GHK-Cu and some other peptides actually downregulate EMT pathways.
Next up: Module 8 — The Brakes Matter More Than the Gas Pedal. p53, PTEN, BRCA1, and the brakes that go missing when cancer takes over.
Module 8 — The Brakes Matter More Than the Gas Pedal
Research and educational purposes only. Not for human consumption.
Why This Module Matters
The first seven modules focused on the gas pedal — the growth and proliferation pathways tumors use to expand. This module is about the brakes. Tumor suppressors are the genes whose normal function is to stop cells from becoming cancerous. When they fail, the gas pedal pathways run unchecked.
Understanding the brakes is what closes the framework. It’s also where the GHK-Cu story gets interesting — because it’s the one peptide in this category whose published gene expression effects actually upregulate tumor suppressors rather than overlapping with pro-growth pathways [13].
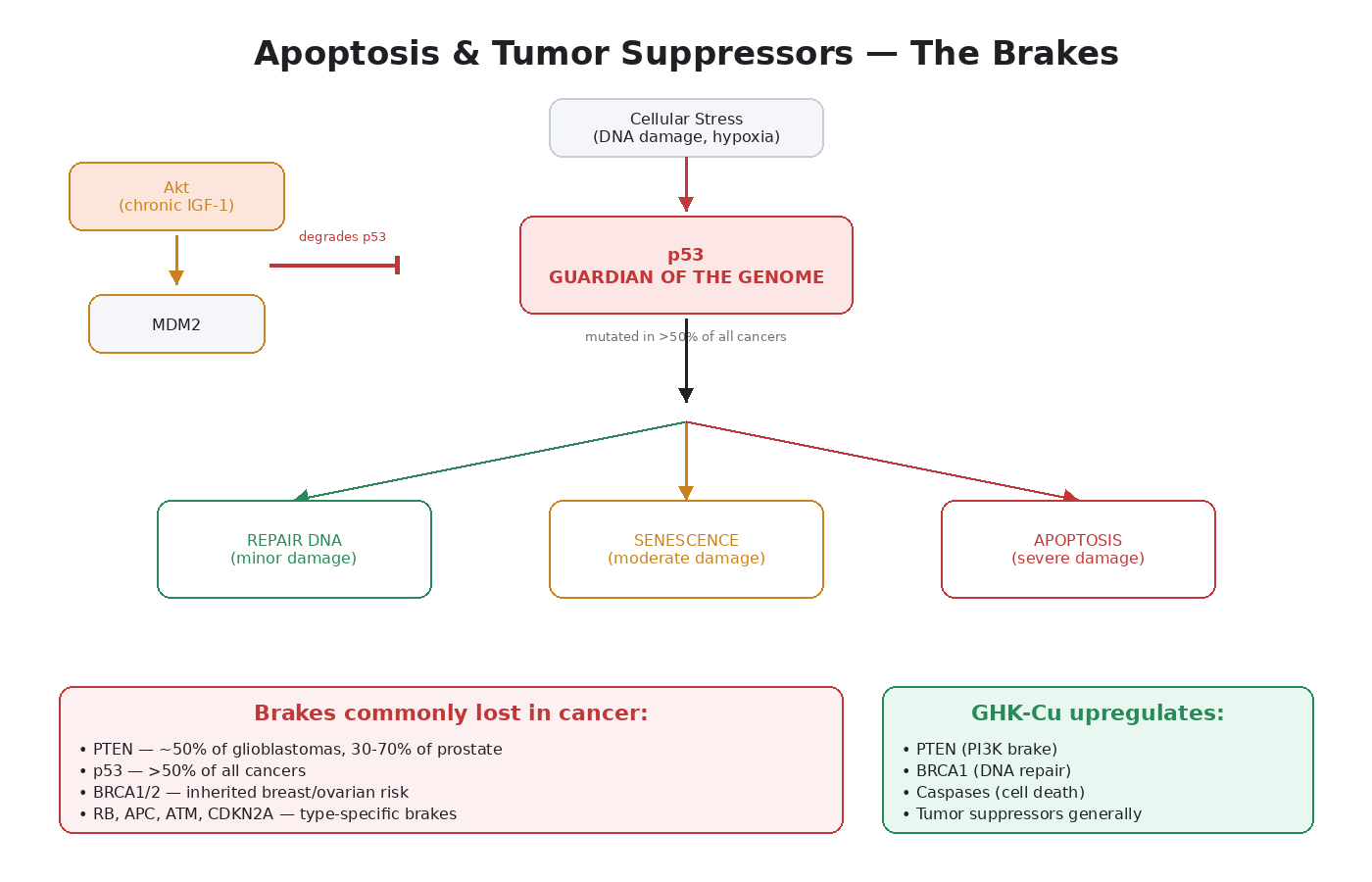
What Apoptosis Is
Apoptosis is programmed cell death — the cell’s built-in self-destruct mechanism. It is fundamentally different from necrosis (uncontrolled cell death from injury). Apoptosis is orderly, regulated, and designed to remove cells without triggering inflammation.
Why does the body need a self-destruct system? Because cells go wrong constantly. Every time a cell divides, there’s a chance of replication errors. Every day, environmental exposures damage DNA. The body’s response to most damaged cells is not to repair them at all costs — it’s to kill them and replace them with fresh ones from the stem cell pool.
When apoptosis works correctly, damaged or potentially dangerous cells are eliminated before they can become a problem. When it fails, those cells survive and accumulate mutations. This is one of the central mechanisms by which cancer develops.
The Apoptosis Machinery
There are two main pathways for triggering apoptosis:
The Intrinsic Pathway (Mitochondrial)
Triggered by internal stress — DNA damage, oxidative stress, growth factor withdrawal, oncogene activation. The key event is permeabilization of the outer mitochondrial membrane.
The control system is the Bcl-2 protein family:
- Pro-apoptotic members (Bax, Bak, Bad, Bid, Bim, PUMA, NOXA) — push toward death
- Anti-apoptotic members (Bcl-2, Bcl-XL, Mcl-1) — push toward survival
The balance between these determines whether the mitochondria release cytochrome c, which activates a cascade of caspases that execute the cell death program.
Tumor cells often overexpress Bcl-2 or downregulate Bax. The drug venetoclax was developed specifically to inhibit Bcl-2 and is approved for several blood cancers.
The Extrinsic Pathway (Death Receptor)
Triggered by external signals — typically immune cells releasing death ligands like FasL or TRAIL that bind death receptors on the target cell surface. This is how cytotoxic T-cells kill virus-infected cells and tumor cells.
Tumor cells often downregulate death receptors or upregulate decoy receptors that absorb the death signals without transmitting them.
How Growth Signaling Inhibits Apoptosis
This is the connection point back to Modules 4 and 5.
When PI3K/Akt is activated:
- Akt phosphorylates BAD (a pro-apoptotic protein), inactivating it.
- Akt phosphorylates and inactivates FOXO transcription factors, which would otherwise activate pro-apoptotic genes.
- Akt activates NF-κB, which drives expression of anti-apoptotic Bcl-2 family members.
- Akt phosphorylates MDM2, accelerating p53 degradation (we’ll get to p53 in a moment).
When MAPK/ERK is activated:
- ERK phosphorylates BIM (a pro-apoptotic protein), targeting it for degradation.
- ERK upregulates Mcl-1 (anti-apoptotic).
- ERK contributes to NF-κB activation.
The net effect: chronic growth signaling is anti-apoptotic. A cell with constantly elevated PI3K/Akt or MAPK/ERK activity is harder to kill. This is good in muscle tissue you’re trying to build. It’s bad in a cell that has accumulated mutations and should be eliminated.
p53: The Guardian of the Genome
p53 is the single most important tumor suppressor in the human body. It is mutated in over 50 percent of all human cancers, making it the most frequently mutated gene in oncology [7].
Normal function: p53 acts as a sensor for cellular stress. When DNA damage occurs, when oncogenes activate inappropriately, or when oxygen levels drop dangerously, p53 levels rise. p53 then makes a decision:
- If the damage is minor — pause the cell cycle and allow DNA repair.
- If the damage is moderate — trigger senescence (permanent cell cycle arrest).
- If the damage is severe — trigger apoptosis.
This decision-making capacity is why p53 is called “the guardian of the genome.” It’s the protein that decides whether a damaged cell gets repaired, retired, or killed.
How tumors disable p53:
- TP53 mutations — the gene itself gets mutated, producing nonfunctional protein. Most common in lung cancer, colorectal cancer, ovarian cancer, esophageal cancer, head and neck cancers.
- MDM2 overexpression — MDM2 is the protein that tags p53 for degradation. Some tumors amplify MDM2, lowering p53 levels without mutating the gene.
- Akt-mediated MDM2 activation — and this is the part that connects to our peptide discussion. When Akt is chronically active, it phosphorylates MDM2, which then degrades p53.
This last mechanism is critical to understand. IGF-1 LR3, by chronically activating PI3K/Akt, indirectly reduces p53 protein levels. Even in cells with normal TP53 genes, sustained Akt signaling lowers the body’s most important cancer brake.
PTEN: The Brake on the Master Pathway
PTEN (Phosphatase and Tensin Homolog) is the main negative regulator of PI3K/Akt/mTOR. As covered in Module 4, PTEN’s job is to convert PIP3 back to PIP2, shutting down the pathway.
PTEN is one of the most frequently lost tumor suppressors in human cancer:
- ~50 percent of glioblastomas
- ~30 to 70 percent of prostate cancers (loss increases with disease progression)
- ~30 percent of endometrial cancers
- ~25 percent of breast cancers
- High percentages in melanoma, lung cancer, colorectal cancer
PTEN loss leaves PI3K/Akt/mTOR constitutively active. The cell receives a “grow and survive” signal regardless of whether external growth factors are present.
Inherited PTEN mutations cause Cowden syndrome, which dramatically increases lifetime cancer risk across multiple organ systems.
The notable peptide connection: GHK-Cu has been shown to upregulate PTEN expression in human breast cancer (MCF7) and prostate cancer (PC3) cell lines, per the Pickart gene expression work [13]. We get into this in detail in Module 12.
BRCA1 and BRCA2: DNA Repair and Inherited Risk
BRCA1 and BRCA2 are tumor suppressors that function in DNA damage repair, specifically homologous recombination repair of double-strand DNA breaks.
When BRCA1/2 are functional, DNA breaks get repaired accurately. When they’re mutated:
- DNA breaks get repaired through error-prone backup mechanisms.
- Mutations accumulate faster.
- Genome instability increases.
- Risk of breast, ovarian, prostate, and pancreatic cancer rises dramatically.
BRCA1 mutation carriers have ~60-70 percent lifetime risk of breast cancer and ~40-50 percent risk of ovarian cancer. BRCA2 has slightly lower but still high lifetime risks.
This is why family history is one of the most important inputs in peptide risk stratification. A user with a known BRCA mutation has compromised DNA repair to start. Adding broad growth signaling to that baseline creates a different risk picture than the same use in someone with intact DNA repair.
GHK-Cu has been shown to upregulate BRCA1 expression in the gene expression studies — another piece of why its profile sits differently than IGF-1 LR3 [13].
Other Important Tumor Suppressors
A few worth knowing:
- RB (Retinoblastoma) — controls the G1-to-S phase transition in the cell cycle. Lost in retinoblastoma (obviously) but also frequently inactivated in many other cancers, often through pathway changes rather than direct mutation.
- APC (Adenomatous Polyposis Coli) — regulates Wnt signaling. Mutated in nearly all colorectal cancers and is the gene behind familial adenomatous polyposis.
- ATM (Ataxia Telangiectasia Mutated) — DNA damage sensor that activates p53. Mutation increases cancer risk.
- CDKN2A (p16) — cell cycle inhibitor. Frequently lost in melanoma and pancreatic cancer.
- VHL (Von Hippel-Lindau) — regulates HIF-1alpha. Mutated in clear cell renal cell carcinoma.
Each of these is a brake. Each one is lost or impaired in specific cancer types. Each one, when functioning, contributes to keeping cells out of the cancer state.
The Bigger Picture: Why Brakes Matter More Than Gas Pedals for Risk Stratification
A healthy person with intact tumor suppressors can tolerate substantial growth signaling without developing cancer. The brakes catch damaged cells before they progress.
A person with compromised brakes — BRCA mutation, PTEN loss, family history of TP53-related syndromes — has much less margin for error. The same level of growth signaling that produces only healing in a brake-intact person can theoretically accelerate progression in someone whose brakes are partially missing.
This is why family history matters so much. The presence of a strong family cancer history doesn’t just indicate higher baseline risk. It specifically suggests that one or more tumor suppressor systems may already be partially compromised. Growth signaling lands differently in that biological context.
The practical implication: peptide risk stratification isn’t just about which compound you’re using. It’s about the biological context the compound is landing in. Brake integrity is at least as important as gas pedal intensity.
Key Takeaways
- Apoptosis is programmed cell death — the body’s quality control system for damaged or dangerous cells.
- The Bcl-2 family controls the intrinsic apoptosis pathway; death receptors control the extrinsic one.
- Tumors resist apoptosis by overexpressing anti-apoptotic proteins or downregulating pro-apoptotic ones.
- Chronic PI3K/Akt and MAPK/ERK activation is anti-apoptotic — this is part of the cancer-pathway overlap of growth peptides.
- p53 is the most important tumor suppressor — mutated in >50% of all cancers.
- Akt-mediated MDM2 activation indirectly reduces p53 — meaning chronic IGF-1 signaling lowers p53 even without TP53 mutations.
- PTEN is the main brake on PI3K/Akt/mTOR — lost in many cancers.
- GHK-Cu uniquely upregulates PTEN, BRCA1, and other tumor suppressors in published cell line studies.
- BRCA1/2 are DNA repair genes — inherited mutations dramatically increase cancer risk.
- Brake integrity matters more than gas pedal intensity for risk stratification.
- Family history isn’t just a statistical marker — it suggests specific tumor suppressor systems may be compromised.
This concludes Volume 1 of the course. Volume 2 picks up with peptide-specific deep dives — BPC-157, TB-500, IGF-1 LR3, GHK-Cu, growth hormone secretagogues, the GHRH peptides, and the practical layer of risk stratification, washout strategy, and where the research is heading.
Module 9 — BPC-157: Healing Through the Same Door Tumors Use
Research and educational purposes only. Not for human consumption.
Why This Module Exists
Modules 1 through 8 built the foundation. Now we apply it to specific peptides.
BPC-157 is the most-discussed regenerative peptide in this community. It has 36+ published preclinical studies, one human trial, and a reputation for healing gut, tendon, ligament, and connective tissue injuries that conventional treatments struggle with [1]. It is also one of the peptides most often asked about in the cancer-risk conversation.
This module is the synthesis: what BPC-157 actually does, where the cancer overlap is, where the nuances live, and how to think about it using the framework from Volume 1.
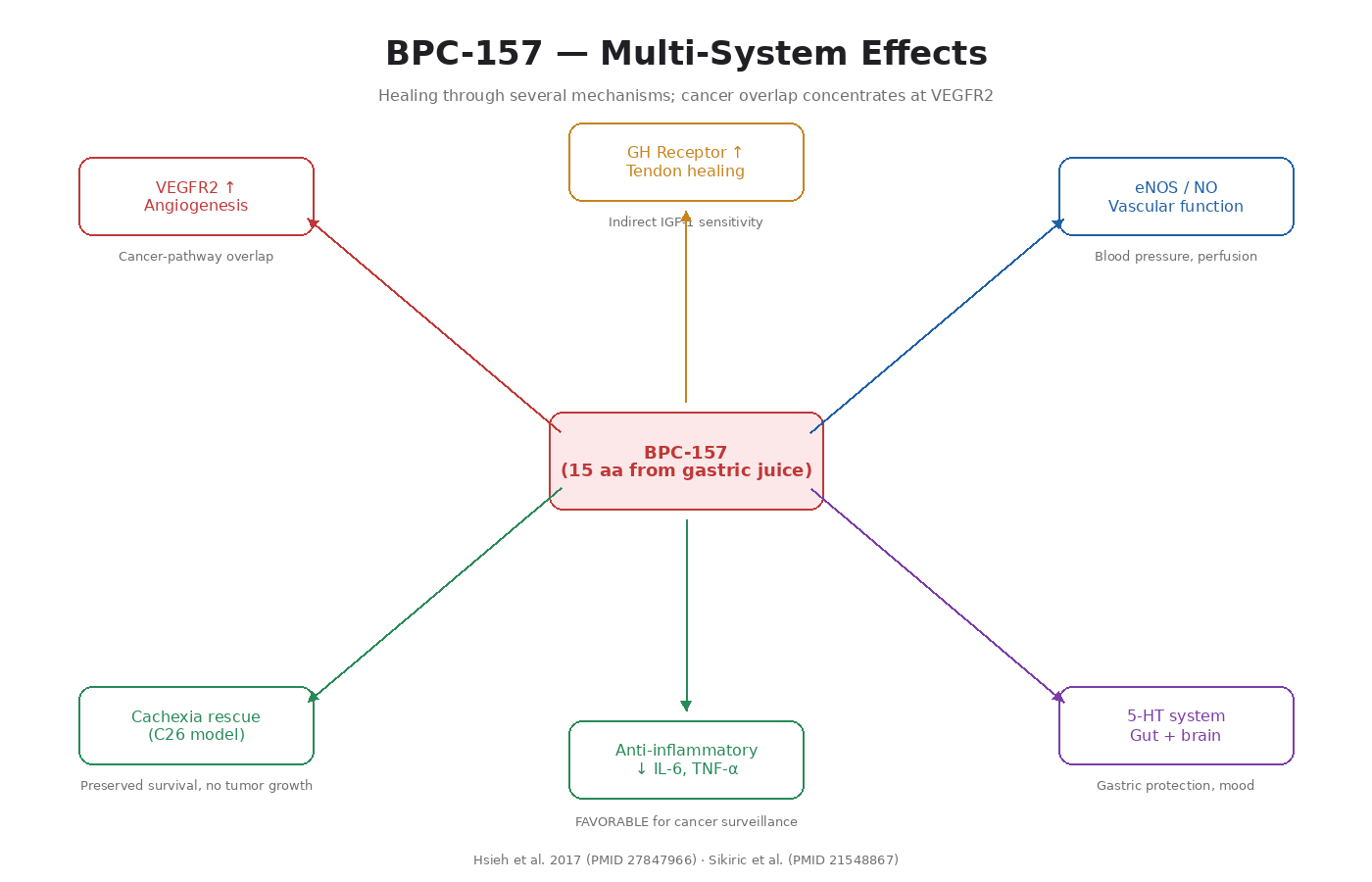
What BPC-157 Is
BPC-157 stands for “Body Protection Compound 157.” It is a synthetic pentadecapeptide — 15 amino acids long — derived from a sequence found in human gastric juice. Researchers first identified it while studying how the stomach protects itself. Since then, it has been shown to drive healing across many tissues [2].
The sequence is GEPPPGKPADDAGLV. Researchers have shown it stays stable in gastric acid, which is unusual for a peptide. That stability is why both oral and injectable routes appear to work in animal studies.
The Documented Mechanisms
BPC-157 affects multiple pathways at once. The well-characterized ones:
1. VEGFR2 Upregulation and Angiogenesis
The most rigorous mechanistic work is Hsieh et al. 2017 [3]. BPC-157:
- Increases VEGFR2 expression on vascular endothelial cells
- Promotes VEGFR2 internalization (required for full signaling)
- Activates the VEGFR2-Akt-eNOS pathway
- Increases endothelial tube formation in cell culture
- Speeds up blood flow recovery in hind limb ischemia models
- Drives angiogenesis in chick chorioallantoic membrane assays
This is the core mechanism behind BPC-157’s healing effects. Better vascularization → better tissue perfusion → faster healing.
2. Growth Hormone Receptor Upregulation
BPC-157 has been shown to upregulate growth hormone receptor expression in tendon fibroblasts [4]. This matters because GH and IGF-1 signaling drive tendon healing. BPC-157 makes tissues more responsive to the body’s existing GH/IGF-1 signaling without raising those hormones systemically.
3. Nitric Oxide System Modulation
BPC-157 interacts with the NO system in complex ways. It maintains and protects eNOS activity, drives NO production through the VEGFR2 pathway, and counteracts both deficient and excessive NO conditions depending on context. That’s why it has effects on blood pressure, vascular function, and even some neurological outcomes.
4. 5-HT (Serotonin) System Effects
BPC-157 modulates serotonergic signaling in the gut and brain. This contributes to its effects on gastric protection, gut motility, and possibly some mood-related outcomes in animal models.
5. Anti-Inflammatory Effects
BPC-157 reduces pro-inflammatory cytokines (IL-6, TNF-alpha) in multiple inflammation models. From the cancer-pathway perspective, this is favorable — chronic inflammation is itself a major cancer driver (Volume 1, Module 2).
6. Counteracts Tumor Cachexia
In a mouse C26 colon adenocarcinoma model, BPC-157 counteracted muscle wasting, preserved myogenesis, prolonged survival, and reduced pro-cachectic cytokines [5]. Importantly, this happened without accelerating tumor growth.
Where the Cancer-Pathway Overlap Is
Applying the Volume 1 framework:
VEGF/VEGFR2 (V1 Module 3): Direct overlap. BPC-157 upregulates the same receptor tumors hijack for angiogenesis. This is the most concerning mechanism in the cancer context.
PI3K/Akt/mTOR (V1 Module 4): Indirect overlap through VEGFR2-Akt signaling. BPC-157 activates Akt as part of its angiogenic effect, which means it contributes to anti-apoptotic and growth signaling in the same cells where VEGFR2 is being activated.
MAPK/ERK (V1 Module 5): Context-dependent. In melanoma cell lines, BPC-157 actually inhibits VEGF signaling via MAPK [6]. In other contexts, it may activate MAPK as part of healing signaling. The data is mixed.
IGF-1R (V1 Module 6): Minimal direct interaction. BPC-157 doesn’t bind IGF-1R or significantly elevate systemic IGF-1.
EMT (V1 Module 7): Indirect through angiogenic and PI3K/Akt signaling, but no direct evidence of EMT promotion. Not in the same risk category as TB-500.
Tumor suppressors (V1 Module 8): No documented effects on p53, PTEN, BRCA1, or other major suppressors. Neutral here.
Where the Anti-Tumor Evidence Is
This is the part that gets underdiscussed. BPC-157 has shown anti-tumor effects in specific preclinical contexts:
- In human melanoma cell lines, BPC-157 inhibited cell growth and VEGF signaling through MAPK kinase pathway modulation [6].
- In the C26 colon adenocarcinoma mouse model, BPC-157 counteracted cachexia and prolonged survival without accelerating tumor growth [5].
- Unpublished mentions of reduced lung metastases in B-16 melanoma models (we should be skeptical of unpublished data, but it’s in the literature reviews).
- Folkman-style corneal neovascularization work: BPC-157 inhibited corneal neovascularization in some contexts, which the original authors connected to potential anti-tumor angiogenic effects.
The picture is genuinely more nuanced than “BPC-157 = pro-tumor angiogenesis.” The peptide appears to have context-dependent effects on angiogenic signaling — driving it in healing contexts and modulating it (sometimes inhibiting it) in tumor contexts.
That said, the conservative position is to treat the mechanistic risk seriously regardless. We have very strong data on VEGFR2 upregulation. We have weaker, more variable data on anti-tumor effects. The default assumption should follow the strongest data.
The Practical Risk Profile
Pulling together what we know:
Lower-risk profile use of BPC-157:
- Younger researchers (under ~40)
- No personal cancer history
- No first-degree family history of cancer
- Clean baseline screening
- Time-limited cycles (4–6 weeks on, 4–6 weeks off)
- Specific healing application (injury-driven use, not chronic prophylactic use)
Higher-risk profile considerations:
- Personal cancer history
- First-degree family history, especially of vascularized solid tumors (colorectal, breast, lung, renal)
- Age above ~50, where subclinical neoplasms become statistically more likely
- Chronic uninterrupted use without washouts
- Stacking with other angiogenic or growth-signaling peptides
This isn’t a permission list. It’s a risk-stratification framework. The mechanism of BPC-157 doesn’t change based on who’s using it — what changes is the baseline biological context the mechanism is landing in.
Comparison to Anti-Angiogenic Cancer Drugs
A useful thought experiment. Bevacizumab (Avastin) is a monoclonal antibody that binds VEGF and prevents it from activating VEGFR2. It’s approved for colorectal, lung, kidney, ovarian, and several other cancers. It works because shutting down angiogenesis starves tumors.
BPC-157 does roughly the opposite at the mechanistic level — it amplifies VEGFR2 signaling. The therapeutic effects in healing are real and well-documented. But the mechanism is, in the most literal sense, the opposite of a major anti-cancer drug class.
This doesn’t mean BPC-157 is dangerous in a healthy body. It means the mechanism deserves the kind of respect you’d give to operating on the same pathway that an anti-cancer drug specifically targets.
Stacking and Pathway Compounding
BPC-157 is often stacked with TB-500. From the framework we’ve built, that stack:
- Amplifies angiogenesis (both compounds, different mechanisms)
- Combines BPC-157’s VEGFR2 sensitization with TB-500’s increased VEGF production
- Adds TB-500’s cell migration effects on top of BPC-157’s tissue-perfusion effects
The healing rationale is sound — the combination addresses multiple healing mechanisms. The cancer-pathway compounding is also real. Stacking these two amplifies pathway activation more than using either alone.
The conservative position: if you’re going to stack, do it for time-limited healing application, not chronic use. The mechanism of action and the metastatic-cascade machinery overlap is something you don’t want chronically running.
The Honest Bottom Line
BPC-157 is one of the most promising regenerative peptides in the literature. The healing data is real. The mechanism is well-characterized. The anti-tumor data in specific cancer models is genuinely interesting and gets less attention than it deserves.
It’s also a peptide that operates on the angiogenic pathway tumors most directly hijack, and that pathway upregulation is its core mechanism, not an off-target effect.
My read of the literature: BPC-157 does not cause cancer. There is no published evidence that exogenous BPC-157 transforms a healthy cell into a cancer cell. But it operates on a pathway that, in a body with an existing or undetected malignancy, could theoretically accelerate progression.
That makes it a peptide where the mechanism deserves real respect, the use case deserves to be specific and time-limited, and the baseline biological context — age, family history, screening status — matters more than it does for compounds without this kind of pathway overlap.
Key Takeaways
- BPC-157 is a 15-amino-acid synthetic peptide derived from a sequence found in gastric juice.
- Its core mechanism is VEGFR2 upregulation, driving angiogenesis and tissue perfusion.
- It also upregulates growth hormone receptor expression, modulates NO and serotonin systems, and reduces pro-inflammatory cytokines.
- The cancer-pathway overlap is most direct at VEGFR2 (V1 Module 3) and indirect through Akt activation (V1 Module 4).
- It has shown anti-tumor effects in specific preclinical contexts — melanoma cell lines, C26 colon adenocarcinoma cachexia models.
- The picture is more nuanced than “BPC-157 = pro-tumor angiogenesis,” but the conservative position respects the mechanistic risk.
- Risk stratification matters: age, family history, screening status, cycle length, stacking.
- Stacking with TB-500 amplifies angiogenic pathway activation more than either alone.
- My read: BPC-157 does not cause cancer, but operates on pathways where undetected malignancy could theoretically be accelerated.
Next up: Module 10 — TB-500. The peptide with the most direct EMT-machinery overlap of anything in this course.
Module 10 — TB-500: The Migration Peptide and the Metastasis Question
Research and educational purposes only. Not for human consumption.
Why This Module Exists
TB-500 deserves its own deep dive because the cancer-pathway overlap here is more direct than for any other peptide in this category. While BPC-157 touches angiogenesis through one specific mechanism (VEGFR2 sensitization), TB-500 touches multiple cancer-relevant pathways at once: cell migration, angiogenesis, EMT signaling, and integrin-linked kinase activation.
The endogenous version of TB-500 — thymosin beta-4 (TB4) — is overexpressed in essentially every aggressive cancer type studied. That doesn’t mean TB-500 causes cancer, but it does mean the mechanistic overlap deserves the most careful evaluation in this course.
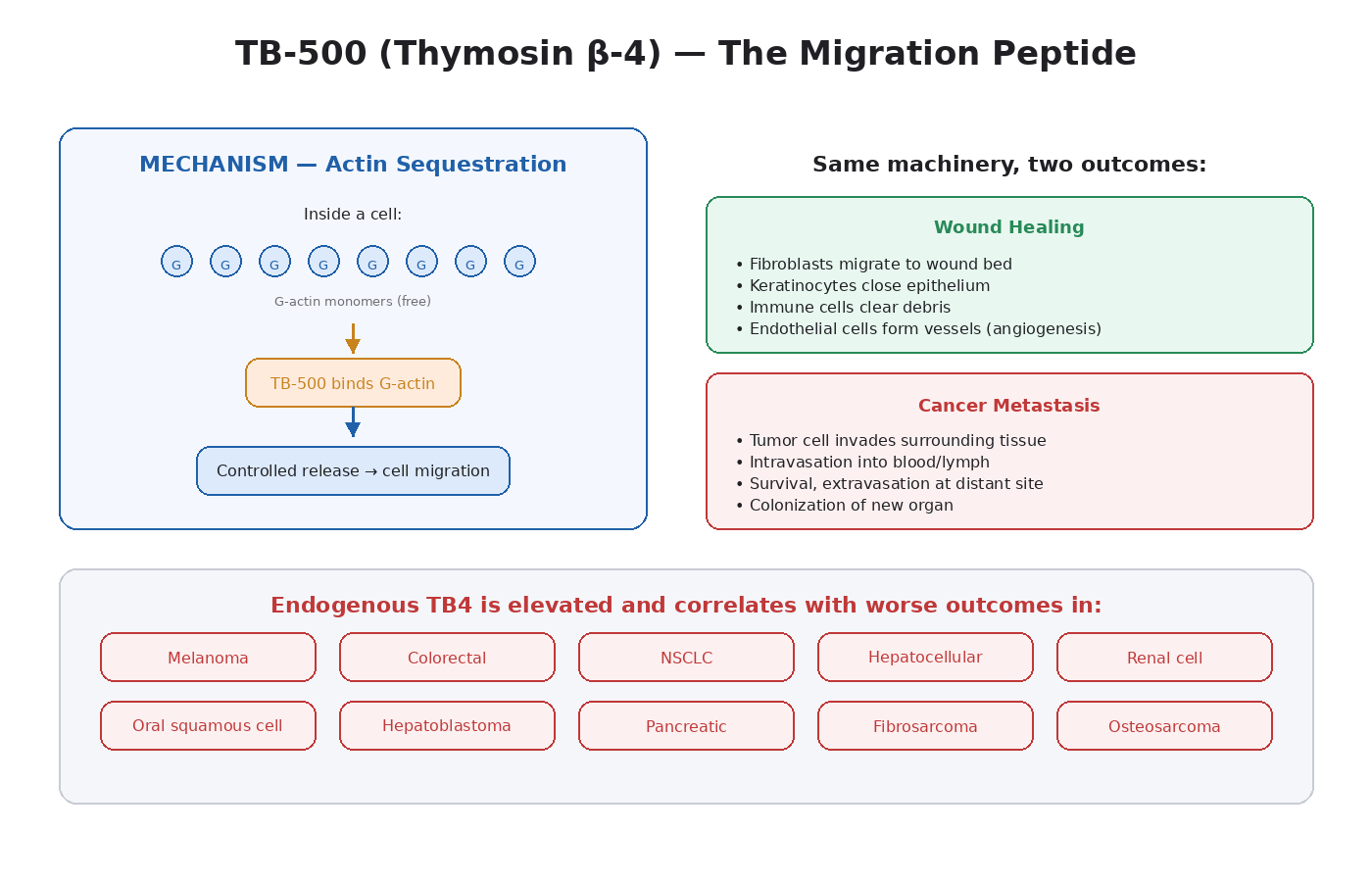
What TB-500 Is
TB-500 is a synthetic fragment of thymosin beta-4 (TB4), a naturally occurring 43-amino-acid peptide that is one of the most abundant intracellular peptides in the human body. It is found in virtually every cell type except red blood cells [7].
The full TB4 sequence is 43 amino acids. TB-500 specifically is a 17-amino-acid fragment (LKKTETQ being the most active core). The synthetic version was developed because the smaller fragment keeps most of TB4’s biological activity while being easier and cheaper to manufacture.
Some commercial “TB-500” products are actually the full-length TB4 peptide. Others are the LKKTETQ fragment. The functional effects are similar enough that the community generally treats them interchangeably, though purity and exact sequence matter for research purposes.
The Documented Mechanisms
1. Actin Sequestration
This is TB4’s foundational function. Inside a cell, actin exists in two forms: G-actin (single monomers floating freely) and F-actin (long filaments that make up the cytoskeleton). TB4 binds G-actin and prevents it from polymerizing into F-actin too early.
This gives the cell a controlled reserve of actin monomers that can be released and polymerized when needed for shape changes, migration, or division. TB4 is one of the most important regulators of cytoskeletal dynamics in the entire body.
2. Cell Migration
By controlling actin polymerization, TB4 enables efficient cell migration. Cells move by extending protrusions (lamellipodia, filopodia) at their leading edge, which requires rapid, localized actin polymerization. TB4 supplies the monomer pool that makes this possible.
In wound healing, this is excellent. Fibroblasts, keratinocytes, and immune cells need to migrate into the wound bed. In cancer, the same machinery enables metastasis.
3. Angiogenesis
TB4 drives angiogenesis through several mechanisms [8]:
- Upregulates VEGF expression in a HIF-1alpha-dependent manner (different from BPC-157, which upregulates VEGFR2 instead)
- Promotes endothelial cell migration and tube formation
- Activates the FAK-paxillin pathway for endothelial cytoskeletal remodeling
4. Anti-Inflammatory Effects
TB4 inhibits NF-κB activation, reduces pro-inflammatory cytokine production, and modulates innate immune responses. From the cancer-pathway perspective, this is favorable.
5. ILK (Integrin-Linked Kinase) Pathway Activation
TB4 activates ILK, which sits at the interface between integrin-mediated cell adhesion signals and intracellular pathways including PI3K/Akt. ILK is itself associated with EMT and cancer progression in the literature.
6. Anti-Apoptotic Effects
TB4 increases cell survival under stress conditions through multiple mechanisms including Akt activation. Good for healing tissue. Less good if expressed in cells that should be apoptosing.
7. Tissue Regeneration Across Organ Systems
TB4 has documented regenerative effects on cardiac tissue (post-MI in rodent models) [8], corneal epithelium, skin wounds, neural tissue, and musculoskeletal injuries.
The Cancer Literature on Endogenous TB4
This is where the conversation has to be unflinching. Endogenous TB4 expression is studied in oncology, and the findings are consistent [9]:
- Melanoma — TB4 is upregulated and correlates with metastatic potential.
- Colorectal cancer — TB4 overexpression correlates with tumor stage and worse outcomes.
- Non-small-cell lung cancer (NSCLC) — elevated TB4 is associated with poor prognosis.
- Hepatocellular carcinoma — TB4 drives invasion and is overexpressed in metastatic disease.
- Renal cell carcinoma — TB4 expression correlates with tumor aggressiveness.
- Oral squamous cell carcinoma — TB4 induces proliferation, invasion, and full EMT.
- Hepatoblastoma — TB4 directly drives EMT through E-cadherin downregulation.
- Pancreatic cancer — TB4 is overexpressed and stimulates pro-inflammatory cytokine release.
- Murine fibrosarcoma — TB4 regulates motility and metastasis.
- Rat osteosarcoma — TB4 contributes to invasive behavior.
The pattern is uniform. Across multiple independent studies, in different cancer types, endogenous TB4 is consistently elevated in aggressive disease and correlates with worse outcomes.
The Important Mechanistic Nuance
TB4 appears to be a driver of cancer aggressiveness, not a transformative agent. This distinction matters and is the same one we drew in Volume 1, Module 1.
There is no published evidence that exogenous TB-500 transforms healthy cells into cancer cells. Cancer cells upregulate TB4 because they need its actin-sequestering, migration-promoting, angiogenic, and anti-apoptotic functions. TB4 is not a mutagen, doesn’t damage DNA, and doesn’t activate oncogenes through mutation.
What the data does show: in a cell that has already acquired oncogenic mutations, TB4 contributes meaningfully to the cell’s ability to migrate, invade surrounding tissue, recruit blood supply, and resist death. These are exactly the capabilities the Hallmarks of Cancer framework (Volume 1, Module 2) identifies as critical for tumor progression.
So the framework holds. TB-500 doesn’t cause cancer. But it operates on machinery aggressive tumors specifically rely on.
Why TB-500 Is Different From BPC-157
Both peptides drive angiogenesis. Both touch growth signaling pathways. Why does TB-500 deserve more cautious framing?
BPC-157 primarily upregulates receptor sensitivity (VEGFR2). It also has documented anti-tumor effects in specific cell line studies. The melanoma VEGF inhibition data is real. The C26 cachexia model shows preserved survival without accelerated tumor growth.
TB-500 primarily drives cell migration. There is no analog to the BPC-157 anti-tumor data — every cancer study on endogenous TB4 shows correlation with worse outcomes. The mechanism is the central machinery of metastasis.
Plus, TB-500 hits multiple cancer-pathway nodes simultaneously:
- Angiogenesis (VEGF/HIF-1alpha)
- Cell migration (the EMT execution mechanism)
- ILK pathway (linked to EMT)
- Anti-apoptotic signaling (Akt activation)
- Anti-inflammatory effects (which can either help or hinder cancer surveillance depending on context)
This combination is mechanistically broader than BPC-157’s profile.
The Practical Risk Profile
Applying the framework:
Lower-risk profile use of TB-500:
- Younger researchers (under ~35)
- Acute, specific musculoskeletal injury indication
- No personal or family cancer history
- Time-limited cycles (4 weeks maximum, with extended washouts)
- Clean baseline screening
Higher-risk profile, with much more careful consideration:
- Personal cancer history of any kind
- First-degree family history of cancer
- Age above ~45
- Chronic uninterrupted use
- Stacking with other growth-signaling or angiogenic peptides
- Active or recent inflammatory conditions in tissues prone to malignancy
The conservative position: TB-500 is a peptide where the cycling and washout discussion matters more than for almost anything else in the regenerative peptide category. Brief, targeted use for a specific healing application is one risk profile. Chronic recreational use for general “anti-aging” or sustained recovery is a different and harder-to-justify risk profile.
The Stacking Question, Revisited
When TB-500 and BPC-157 are stacked, the angiogenic and pro-migratory pathway activation compounds. From a healing standpoint, this is part of why the combination is so well-regarded — it covers multiple healing mechanisms.
From the cancer-pathway standpoint:
- VEGF production goes up (TB-500) AND VEGFR2 sensitivity goes up (BPC-157).
- Cell migration machinery is amplified (TB-500) on top of improved tissue perfusion (BPC-157).
- Akt activation is hit from multiple inputs.
This stack is fine for acute, time-limited healing of a specific injury in a low-risk-profile researcher. It is not a stack to run chronically.
What the Community Often Gets Wrong About TB-500
Two recurring framings that don’t match the literature:
“TB-500 is just a healing peptide, no risks.” This understates the cancer-pathway overlap. The oncology data on endogenous TB4 is consistent and substantial.
“TB-500 causes cancer.” This overstates what we know. No human study has shown exogenous TB-500 administration causing malignancy in healthy subjects. The mechanism is pathway overlap with existing cancer cells, not initiation.
The honest middle position: TB-500 is a peptide with real healing utility and a real cancer-pathway overlap. The mechanism deserves respect, the use case should be specific, and the cycling discipline matters.
The Honest Bottom Line
TB-500 is the regenerative peptide with the most direct overlap with cancer machinery in this entire course. Endogenous TB4 is consistently elevated in aggressive cancers and drives EMT, invasion, angiogenesis, and survival.
My read of the literature: TB-500 does not cause cancer. There is no evidence it transforms healthy cells. But it operates on the exact machinery aggressive tumors depend on, and the mechanism overlap is broader than any other peptide we discuss.
The practical implication: TB-500 is a peptide for acute, specific, time-limited applications. It is not a chronic “stack” component. The cycling discussion matters more here than almost anywhere else. The baseline biological context — age, family history, screening — matters more here than for compounds without this depth of cancer-pathway involvement.
Key Takeaways
- TB-500 is a synthetic fragment of thymosin beta-4, one of the most abundant intracellular peptides in the body.
- Core functions: actin sequestration, cell migration, angiogenesis, anti-inflammatory effects, anti-apoptosis.
- Endogenous TB4 is consistently elevated in aggressive cancers across many tumor types.
- TB4 directly drives EMT in multiple cancer models — most directly relevant to V1 Module 7.
- TB-500 doesn’t cause cancer (not a mutagen, doesn’t damage DNA) but operates on the machinery of metastasis.
- More direct cancer-pathway overlap than BPC-157, no equivalent anti-tumor cell-line evidence.
- Stacking with BPC-157 compounds angiogenic and pro-migratory signaling.
- Lower-risk profile: acute injury indication, time-limited, no cancer history, good screening.
- Higher-risk profile demands much more careful consideration.
- This is the peptide where cycling and washout discipline matters most.
Next up: Module 11 — IGF-1 LR3. The peptide with the most documented epidemiological cancer overlap.
Module 11 — IGF-1 LR3: The Peptide With the Loudest Cancer Signal
Research and educational purposes only. Not for human consumption.
Why This Module Exists
IGF-1 LR3 is the peptide where the cancer-pathway overlap is not theoretical, not preclinical-only, and not based on indirect mechanism. It’s epidemiological. It’s documented in clinical oncology literature spanning over two decades. Pharmaceutical companies have spent billions of dollars developing IGF-1R inhibitors specifically as cancer treatments [10].
If you only read one peptide-specific module in this course, this is the one.
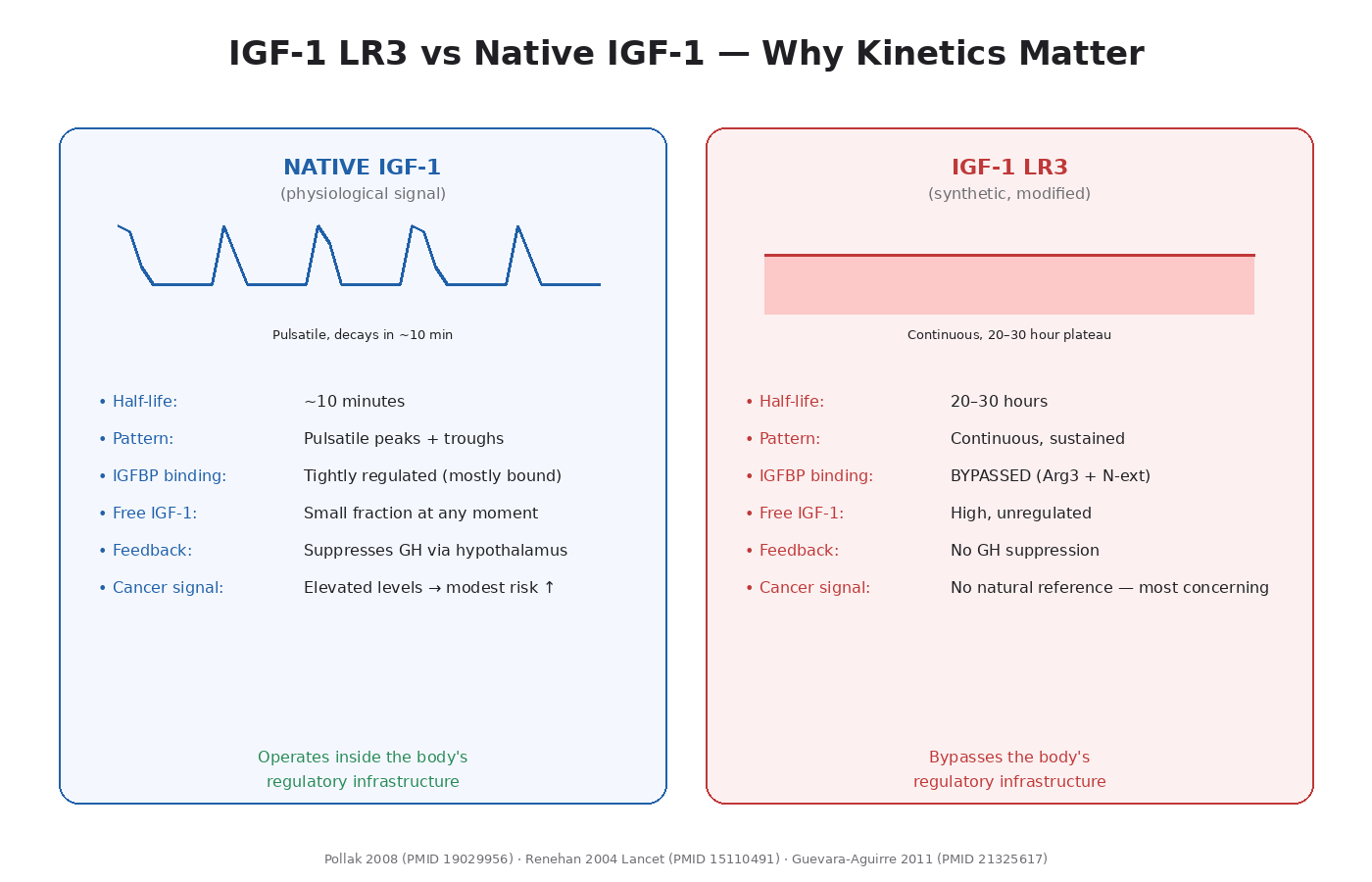
What IGF-1 LR3 Is
Insulin-like Growth Factor 1 Long Arg3 (IGF-1 LR3) is a synthetic analog of native IGF-1 with two modifications:
- Arg3 substitution — the third amino acid is changed from glutamic acid to arginine.
- N-terminal extension — 13 extra amino acids on the front end.
These changes produce two functional effects:
- Dramatically reduced binding to IGF-binding proteins (IGFBPs), particularly IGFBP-3, which normally sequesters most circulating IGF-1.
- Extended half-life: roughly 20–30 hours vs ~10 minutes for native IGF-1.
The net effect is that IGF-1 LR3 produces 2–3 times the bioavailability at IGF-1R and stays in circulation orders of magnitude longer than native IGF-1.
This is exactly what makes it anabolic. It also bypasses one of the body’s main regulatory mechanisms for keeping IGF-1 signaling tightly controlled.
The Mechanism, Step by Step
When IGF-1 LR3 binds IGF-1R:
- The receptor dimerizes and autophosphorylates.
- Adapter proteins (IRS-1, Shc) dock.
- PI3K activates → Akt → mTOR (V1 Module 4).
- MAPK pathway activates in parallel → RAS → RAF → MEK → ERK (V1 Module 5).
- Both pathways drive their full set of downstream effects simultaneously.
The dual pathway activation is the heart of why IGF-1 LR3 is so anabolic. PI3K/Akt drives protein synthesis and anti-apoptotic survival. MAPK/ERK drives cell division and proliferation. Together, they tell cells to grow, divide, and resist death — all at once.
In muscle tissue, this manifests as hypertrophy and satellite cell activation. The peptide community has researched this extensively, and the anabolic effects are well-established.
The Cancer Literature
This is where the conversation has to be unflinching. The IGF-1R cancer literature is one of the most consistent bodies of evidence in oncology:
Epidemiological Evidence
- Elevated circulating IGF-1 is associated with increased risk of colorectal, breast, and prostate cancer across multiple large prospective cohort studies [10].
- A 2004 Lancet meta-analysis pooled the data and confirmed elevated IGF-1 levels correlate with increased risk of prostate, colorectal, and premenopausal breast cancer [11].
- The Physicians’ Health Study showed men in the highest IGF-1 quartile had ~4-fold higher risk of prostate cancer compared to the lowest quartile.
- The IGF-1/IGFBP-3 ratio may be a better predictor than absolute IGF-1 levels.
Laron Syndrome Data
The Ecuadorian Laron syndrome cohort is one of the most striking pieces of evidence in this entire literature [12]. Laron syndrome patients have GH receptor mutations that prevent IGF-1 production. They are very short, but they have:
- Essentially zero rates of cancer in the populations studied
- Dramatically reduced diabetes rates
- Some evidence of extended healthspan
The interpretation: deficient IGF-1 signaling appears to protect against cancer in humans, not just in model organisms.
Acromegaly Data
Acromegaly is the opposite condition — chronically elevated GH and IGF-1 from a pituitary tumor. Acromegalics have:
- Higher rates of colorectal cancer specifically (well-documented)
- Some increased rates of other cancers
- Elevated cancer mortality compared to age-matched controls
This is the closest natural experiment to chronic IGF-1 elevation in adults, and the data is consistent with the epidemiological signal.
Tumor IGF-1R Expression
- Breast cancer: IGF-1R overexpressed in ~50% of cases
- Prostate cancer: high IGF-1R expression in both androgen-dependent and androgen-independent disease
- Colorectal cancer: elevated IGF-1R correlates with worse outcomes
- Lung cancer: IGF-1R overexpression in many cases
- Clear cell renal cell carcinoma: IGF-1R overexpression carries a 70% increased risk of death compared to tumors without IGF-1R expression
- Hepatocellular carcinoma: IGF-1R is a major progression driver
- Ewing sarcoma and other pediatric sarcomas: IGF-1R is essentially required for tumor survival
Drug Development
IGF-1R has been a major pharmaceutical target for over a decade [13]. Drugs developed specifically to block IGF-1R for cancer treatment include:
- Monoclonal antibodies: figitumumab, ganitumab, cixutumumab
- Small molecule inhibitors: linsitinib, BMS-754807
- Multiple combination protocols with mTOR inhibitors
The clinical results have been mixed — IGF-1R blockade alone often isn’t enough because tumors compensate through hybrid receptor signaling with the insulin receptor. But the fact that an entire drug development effort exists to block what IGF-1 LR3 activates is the most direct cancer-pathway overlap in this entire course.
What This Means for LR3 Specifically
IGF-1 LR3 differs from native IGF-1 in two critical ways:
Bypasses IGFBPs. Normal physiology uses IGFBPs to sequester most IGF-1 in circulation. Only a small fraction is biologically active at any moment. LR3 doesn’t engage this regulation. It binds IGF-1R freely.
Sustained signaling. Native IGF-1 has a 10-minute half-life. Circulating levels fluctuate. LR3 produces continuous, elevated signaling for 24+ hours.
The combination — chronic, unregulated, high-amplitude IGF-1R activation — is biologically different from natural elevation. The epidemiological data on IGF-1 and cancer is based on people with naturally elevated but still IGFBP-regulated, pulsatile levels. LR3 use produces a signaling pattern that has no good natural reference and is, mechanistically, the more aggressive version of the same pathway.
The Risk Stratification Reality
Of all the peptides in this course, IGF-1 LR3 is the one where the risk-stratification framework matters most.
Substantial risk considerations:
- Personal history of any cancer, especially hormone-sensitive or epithelial
- First-degree family history of breast, prostate, colorectal, or endometrial cancer
- BRCA1/2 mutations or other inherited cancer predisposition syndromes
- Age above ~40 — subclinical neoplasms become statistically more common
- Active or treated diabetes with hyperinsulinemia (overlapping receptors)
- Acromegaly or any condition with baseline elevated IGF-1
- Chronic colonic adenomas or polyposis (IGF-1R hyperactive context)
Lower-risk profile considerations:
- Younger researchers (under ~30)
- No personal or family cancer history
- Clean baseline screening (including age-appropriate colonoscopy if approaching 40)
- Time-limited, low-dose cycles
- Single specific research application (not chronic general use)
- Long washouts (6+ weeks) between cycles
This isn’t a list of “if these apply, don’t use it.” It’s a list of inputs to the conversation that should be informing the use decision.
The Stacking Question
IGF-1 LR3 is sometimes stacked with growth hormone secretagogues (CJC-1295, ipamorelin, MK-677). From a cancer-pathway perspective, this is the most concerning stack possible — you’re combining direct IGF-1R hyperactivation with elevated endogenous IGF-1 production.
The signaling output is multiplicative, not additive. PI3K/Akt/mTOR is hit harder, longer, and more chronically than either compound alone would produce.
The conservative position: do not stack IGF-1 LR3 with GH secretagogues. If using one, do not use the other in the same cycle. If using both is research-relevant, alternate cycles with substantial washout between them.
The Honest Bottom Line
IGF-1 LR3 is the peptide in the entire research community space where the cancer-pathway overlap is least theoretical. We have epidemiological data, natural experiment data (Laron syndrome, acromegaly), tumor biology data, and active drug development specifically targeting the receptor LR3 activates.
My read of the literature: IGF-1 LR3 does not cause cancer. It is not a mutagen. There is no evidence it transforms healthy cells. But it activates the exact receptor and pathways that aggressive tumors specifically depend on, and it does so with a kinetic profile (chronic, IGFBP-bypassing, sustained) that has no natural physiological analog.
For anyone with cancer history in their family or personal background, this peptide deserves the most careful consideration of anything in this course. For everyone else, the basic discipline — short cycles, long washouts, no stacking with GH secretagogues, baseline screening before use — is the minimum responsible framework.
The honest position is not “don’t use it.” The honest position is: understand exactly what you’re activating, in what biological context, for what specific goal, with what time-limit. If those questions can’t be answered cleanly, the peptide is being used recreationally rather than research-purposefully, and the risk profile shifts accordingly.
Key Takeaways
- IGF-1 LR3 is a synthetic IGF-1 analog with reduced IGFBP binding and extended half-life.
- It activates both PI3K/Akt/mTOR and MAPK/ERK simultaneously — the most potent dual pathway activation in this category.
- The cancer literature is the most consistent in this entire course: elevated IGF-1 correlates with multiple cancer risks.
- Laron syndrome (low IGF-1 signaling) patients have essentially zero cancer rates.
- Acromegaly (high IGF-1) is associated with elevated colorectal cancer specifically.
- IGF-1R is overexpressed in breast, prostate, colorectal, lung, renal (70% increased death risk in ccRCC), and many other cancers.
- Multiple pharmaceutical programs have developed IGF-1R inhibitors as cancer drugs.
- LR3 specifically bypasses IGFBP regulation, producing signaling that has no natural analog.
- Risk stratification matters more here than for any other peptide.
- Do not stack with GH secretagogues.
- The peptide doesn’t cause cancer, but operates on machinery aggressive tumors depend on.
Next up: Module 12 — GHK-Cu. The peptide that breaks the pattern — and why mechanism specificity matters.
Module 12 — GHK-Cu: The Peptide That Breaks the Pattern
Research and educational purposes only. Not for human consumption.
Why This Module Exists
If the last three modules built the case that growth-signaling peptides operate on cancer-pathway machinery, this module is the corrective. Not every peptide in the “regenerative” category follows the same pattern. GHK-Cu has a published gene expression profile that is fundamentally different from BPC-157, TB-500, or IGF-1 LR3 — and understanding this difference is how you stop thinking about peptides categorically and start thinking about them mechanistically [14].
This module is also where the framework gets its most important test. If “peptides feed cancer pathways” were a universal rule, GHK-Cu wouldn’t fit. The fact that it doesn’t fit is what proves the rule is actually about specific mechanisms, not about peptides in general.
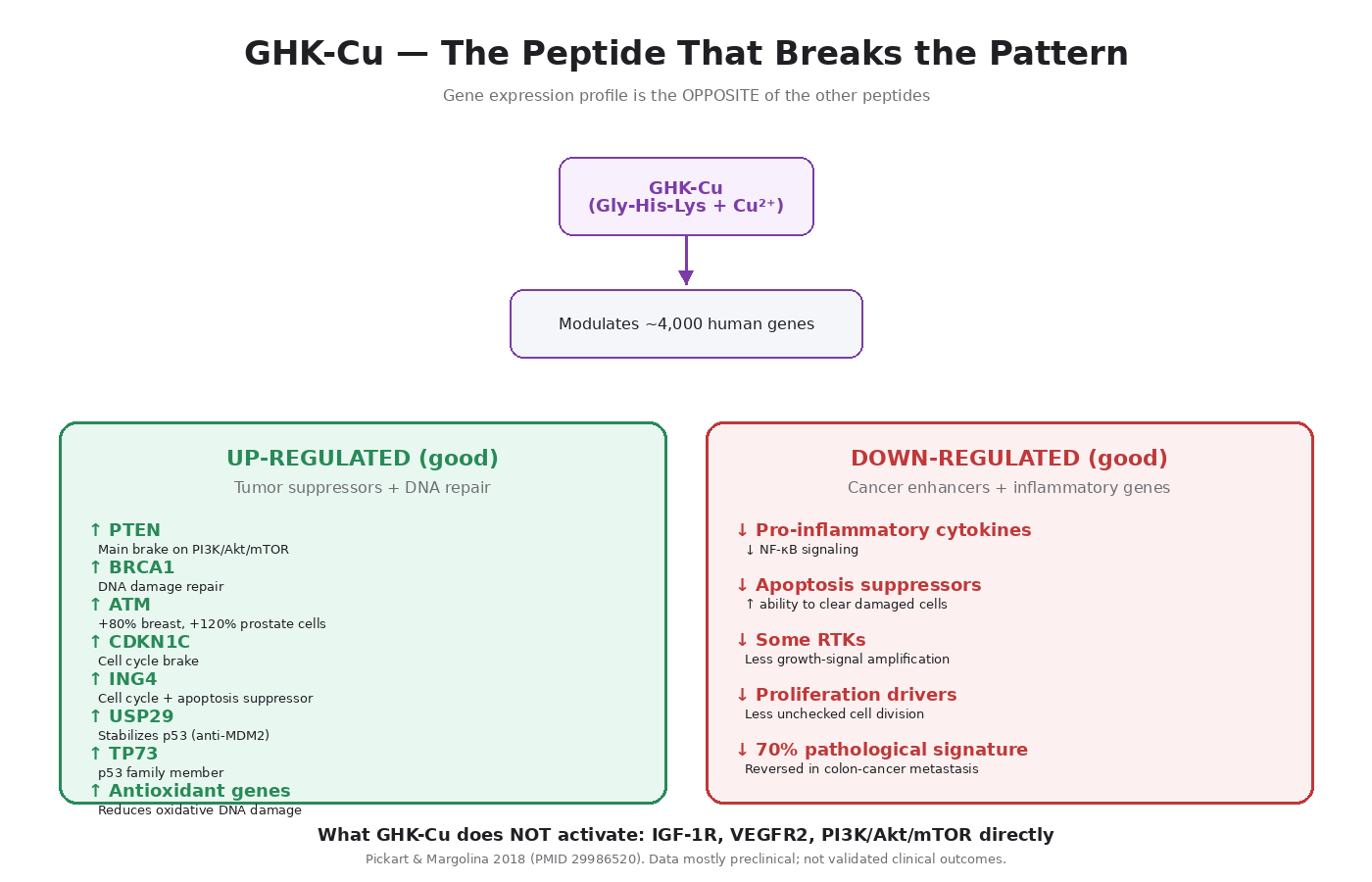
What GHK-Cu Is
GHK is a naturally occurring human tripeptide — just three amino acids: glycyl-L-histidyl-L-lysine. It is found in human plasma at meaningful concentrations, and its levels decline with age (from roughly 200 ng/mL in 20-year-olds to about 80 ng/mL in 60-year-olds).
GHK has a very strong affinity for copper ions. The complex of GHK with copper(II) is called GHK-Cu, and this is the biologically active form most commonly used in research. The copper isn’t a contaminant or additive — it’s an integral part of the molecule’s function.
GHK-Cu was originally identified as a wound-healing factor in serum. It has since been characterized as a regulator of gene expression, a stem cell modulator, an antioxidant, and a tissue regeneration agent. It is also one of the few peptides with substantial clinical use — it’s an established cosmetic ingredient with decades of topical use data.
The Mechanisms — A Different Pattern
GHK-Cu doesn’t operate primarily through receptor binding the way IGF-1 LR3 or BPC-157 do. Its main mechanism is broad gene expression modulation. Pickart and colleagues have published extensively on GHK’s effects on the human genome [14], and the pattern is striking.
In published gene expression analyses, GHK modulates the expression of roughly 4,000 human genes — pushing about a third toward higher expression and a third toward lower expression. The overall pattern reflects a “shift from disease state toward healthy state” across multiple conditions, including COPD and several cancer signatures.
The specific pathways:
1. Tumor Suppressor Upregulation
This is the most cancer-relevant finding. In Pickart’s published studies on MCF7 (human breast cancer) and PC3 (human prostate cancer) cell lines, GHK upregulates multiple tumor suppressors:
- PTEN — the main brake on PI3K/Akt/mTOR (V1 Modules 4 and 8). PTEN is one of the most frequently lost tumor suppressors in cancer.
- BRCA1 — DNA damage repair, inherited cancer syndrome gene.
- CDKN1C — cyclin-dependent kinase inhibitor, cell cycle brake.
- ING4 — tumor suppressor involved in cell cycle and apoptosis.
- C13orf18 and PWR — additional tumor suppressors.
2. p53 Pathway Stabilization
GHK upregulates USP29, which is a ubiquitin-specific peptidase that stabilizes p53 by removing the ubiquitin tags that would otherwise mark it for degradation. This is functionally the opposite of what Akt does (Akt activates MDM2, which tags p53 for degradation).
It also upregulates TP73 (p73), a member of the p53 transcription factor family that has overlapping tumor suppressor functions.
3. DNA Repair Gene Activation
GHK upregulates a substantial set of DNA repair genes, including ATM (the master DNA damage sensor) — 80% increase in breast cancer cells, 120% increase in prostate cancer cells in Pickart’s data.
4. Cancer Enhancer Downregulation
GHK downregulates expression of several genes that are typically upregulated in cancer:
- Genes involved in inflammation and cell proliferation signaling
- Apoptosis suppressors (GHK reduces the brakes on cell death)
- Some receptor tyrosine kinase family members
5. Anti-Inflammatory Effects
GHK reduces NF-κB signaling and pro-inflammatory cytokine production. Given that chronic inflammation is itself a major cancer driver (V1 Module 2), this is another favorable signal.
6. Antioxidant Defense
GHK upregulates antioxidant defense genes, increases levels of antioxidant enzymes, and protects cells from oxidative damage. Oxidative DNA damage is one of the contributors to cancer initiation, so this is again favorable.
7. Anti-Cancer Gene Signature Reversal
The headline finding from Pickart’s work: GHK has been shown to reverse approximately 70 percent of the pathological gene expression in a published metastasis-prone colon cancer gene signature [14]. This isn’t a claim that GHK treats cancer — it’s a gene expression observation. But it’s a striking observation.
What GHK-Cu Doesn’t Do
To be precise about the framework:
- GHK-Cu does not significantly activate IGF-1R.
- It does not directly drive PI3K/Akt/mTOR.
- It does not significantly upregulate VEGFR2 the way BPC-157 does.
- It does not have the cell migration mechanism TB-500 has.
- It does not produce sustained mitogenic signaling.
In other words, the pathways that produce the cancer-pathway overlap in the other peptides are not the pathways GHK-Cu primarily affects.
The Important Caveats
This module is the counter-case, but it has to be honest about limitations.
The data is mostly preclinical. The gene expression studies are in cancer cell lines and bioinformatic analyses. There is no clinical outcome data showing GHK-Cu treats or prevents cancer in humans. The findings are mechanistically interesting, not clinically validated.
Copper biology is complex. Copper itself has dual roles in cancer biology. Some literature suggests that elevated copper can support tumor angiogenesis and metastasis. The GHK-Cu complex appears to behave differently from free copper because the chelation modulates copper’s bioavailability and tissue distribution. But the broader question of copper’s role in cancer is more complicated than the GHK-specific data alone.
Concentration matters. Most of the favorable gene expression effects are observed at specific concentrations of GHK-Cu. Whether systemic administration produces those same intracellular concentrations is not fully characterized.
Anti-cancer cell line data doesn’t equal anti-cancer in humans. Many compounds have shown impressive activity in cell lines and failed in clinical translation. This is a well-known problem in oncology drug development.
The honest framing: GHK-Cu has a fundamentally different mechanistic profile than the other peptides in this category. The pattern of effects — tumor suppressor upregulation, p53 stabilization, DNA repair activation — is genuinely the opposite of what concerns us with IGF-1 LR3 or TB-500. But “different mechanism” is not the same as “cancer-protective.”
Why the Mechanism Difference Matters for Risk Stratification
If you accept the framework that peptides don’t cause cancer but can feed pathways tumors hijack, then the question for any specific peptide becomes: which pathways?
- BPC-157: VEGFR2 sensitization → angiogenesis. Concerning if undetected vascularized tumor.
- TB-500: Cell migration + angiogenesis + ILK pathway → metastasis machinery. Concerning broadly.
- IGF-1 LR3: PI3K/Akt/mTOR + MAPK/ERK direct activation → most documented overlap. Concerning for many cancer contexts.
- GHK-Cu: Tumor suppressor upregulation + DNA repair activation + anti-inflammatory effects. The pathways being modulated are largely the brakes, not the gas pedal.
This is why blanket statements like “all growth peptides increase cancer risk” miss the point. The mechanism specificity determines the risk profile. GHK-Cu sits in a fundamentally different mechanistic category, and the framework can accommodate that without being inconsistent.
Practical Considerations
GHK-Cu’s risk profile, applied:
Generally favorable considerations:
- No documented activation of major cancer-driver pathways
- Tumor suppressor upregulation in published cell line studies
- Anti-inflammatory profile favors cancer surveillance
- Antioxidant effects reduce DNA damage contribution
- Decades of cosmetic use data without elevated cancer reports
- Mechanism is broad gene expression modulation rather than receptor hyperactivation
Still-warranted caution:
- Most data is preclinical
- Copper biology is complex
- Systemic vs topical use are different exposure profiles
- High-dose systemic use hasn’t been characterized as thoroughly as topical
The practical implication: GHK-Cu is probably the lowest-cancer-pathway-risk peptide in the regenerative category — but probably-the-lowest-risk is not the same as zero-risk, and conclusions about long-term systemic use should be drawn cautiously given the limits of available data.
Why This Module Is Critical for the Whole Framework
If you can’t articulate why GHK-Cu is mechanistically different from IGF-1 LR3, you’re treating peptides as a category instead of as specific molecules with specific pathway effects. That category-level thinking is exactly what produces both the dismissive (“peptides don’t cause cancer, period”) and the alarmist (“all peptides feed cancer”) framings.
The accurate framing — that peptides do not cause cancer but specific peptides operate on specific pathways with varying degrees of cancer-machinery overlap — requires understanding the mechanism of each compound. GHK-Cu is the case that makes the framework necessary. Any framework that flattens GHK-Cu and IGF-1 LR3 into the same risk bucket is wrong.
Key Takeaways
- GHK-Cu is a copper-binding tripeptide naturally present in human plasma.
- Its primary mechanism is broad gene expression modulation, not receptor hyperactivation.
- Published studies show GHK upregulates major tumor suppressors: PTEN, BRCA1, CDKN1C, ING4.
- It upregulates USP29, which stabilizes p53 — the opposite of what Akt-MDM2 does.
- It upregulates DNA repair genes, including ATM.
- It downregulates several cancer enhancer genes.
- It reverses ~70% of pathological gene expression in a published colon cancer metastasis signature.
- It does not significantly activate IGF-1R, VEGFR2, or PI3K/Akt/mTOR.
- Most data is preclinical/cell line — not validated clinical outcome data.
- Copper biology has dual roles in cancer; GHK-Cu chelation appears to behave differently from free copper.
- Mechanism-specific thinking is what makes the peptide-cancer framework actually useful.
- GHK-Cu is probably the lowest-cancer-pathway-risk peptide in the regenerative category, with reasonable caveats.
Next up: Module 13 — Growth Hormone Secretagogues. CJC, ipamorelin, MK-677, and how indirect IGF-1 elevation compares to direct LR3 use.
References
- Sikiric P, Seiwerth S, Rucman R, et al. Stable gastric pentadecapeptide BPC 157: novel therapy in gastrointestinal tract. Curr Pharm Des. 2011;17(16):1612–1632. PMID: 21548867.
- Sikiric P, Seiwerth S, Rucman R, et al. Brain-gut axis and pentadecapeptide BPC 157: theoretical and practical implications. Curr Neuropharmacol. 2016;14(8):857–865. PMID: 27138887.
- Hsieh MJ, Liu HT, Wang CN, et al. Therapeutic potential of pro-angiogenic BPC157 is associated with VEGFR2 activation and up-regulation. J Mol Med. 2017;95(3):323–333. PMID: 27847966.
- Chang CH, Tsai WC, Lin MS, Hsu YH, Pang JH. The promoting effect of pentadecapeptide BPC 157 on tendon healing involves tendon outgrowth, cell survival, and cell migration. J Appl Physiol. 2011;110(3):774–780. PMID: 21030672.
- Kang EA, Han YM, An JM, et al. BPC157 as potential agent rescuing from cancer cachexia. Curr Pharm Des. 2018;24(18):1947–1956. PMID: 29512451.
- Vukojević J, Siroglavić M, Kašnik K, et al. Rat inferior caval vein (ICV) ligature and particular new insights with the stable gastric pentadecapeptide BPC 157. Vascul Pharmacol. 2018;106:54–66. PMID: 29550617.
- Goldstein AL, Hannappel E, Sosne G, Kleinman HK. Thymosin β4: a multi-functional regenerative peptide. Basic properties and clinical applications. Expert Opin Biol Ther. 2012;12(1):37–51. PMID: 22074294.
- Smart N, Risebro CA, Melville AAD, et al. Thymosin β4 induces adult epicardial progenitor mobilization and neovascularization. Nature. 2007;445(7124):177–182. PMID: 17108969.
- Sribenja S, Wongkham S, Wongkham C, Yao Q, Chen C. Roles and mechanisms of beta-thymosins in cell migration and cancer metastasis: an update. Cancer Invest. 2013;31(2):103–110. PMID: 23320793.
- Pollak MN. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8(12):915–928. PMID: 19029956.
- Renehan AG, Zwahlen M, Minder C, O’Dwyer ST, Shalet SM, Egger M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: systematic review and meta-regression analysis. Lancet. 2004;363(9418):1346–1353. PMID: 15110491.
- Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med. 2011;3(70):70ra13. PMID: 21325617.
- Chitnis MM, Yuen JS, Protheroe AS, Pollak M, Macaulay VM. The type 1 insulin-like growth factor receptor pathway. Clin Cancer Res. 2008;14(20):6364–6370. PMID: 18927274.
- Pickart L, Margolina A. Regenerative and protective actions of the GHK-Cu peptide in the light of the new gene data. Int J Mol Sci. 2018;19(7):1987. PMID: 29986520.
- Falutz J, Allas S, Blot K, et al. Metabolic effects of a growth hormone-releasing factor in patients with HIV. N Engl J Med. 2007;357(23):2359–2370. PMID: 18057338.