This is the first part of a three-part research-use series on how the major research peptides relate to cancer biology. The framework: peptides do not cause cancer, but several of them work on the same pathways tumors use to grow. Part 1 builds the foundation — what cancer actually is, how it begins, and the five major signaling pathways that almost every peptide-cancer question maps onto.
Parts in the series:
- Part 1 (you are here) — Cancer biology foundations: initiation vs promotion, hallmarks, VEGF/VEGFR2, PI3K/Akt/mTOR, MAPK/ERK, IGF-1R
- Part 2 — EMT, apoptosis, and the peptide deep dives: BPC-157, TB-500, IGF-1 LR3, GHK-Cu
- Part 3 — Growth hormone secretagogues, risk stratification, washout strategy, and peptides being studied as cancer adjuncts
Module 1 — Two Doors, Not One: How Cancer Actually Begins
Research and educational purposes only. Not for human consumption.
Why This Module Comes First
Almost every wrong take in the “do peptides cause cancer” conversation comes from one mistake. People collapse two completely different stages of cancer biology into one bucket.
Once you understand the difference between initiation and promotion, every other module in this course will make sense.
If you only read one module, read this one.
What Cancer Actually Is
Cancer is not a single disease. It is a category of conditions that share one feature: cells that should have stopped dividing, didn’t.
Every cell in your body has a lifecycle. It grows. It does its job. Then it either divides in a controlled way or it kills itself in a controlled way (a process called apoptosis) and gets cleared out.
That lifecycle is run by genes. Two categories matter:
- Oncogenes are the gas pedal. When they are switched on, cells grow and divide.
- Tumor suppressor genes are the brakes. They tell cells to stop dividing, repair damage, or die if the damage is too severe.
A healthy cell has a balanced gas-pedal-and-brake system. A cancer cell has the gas stuck down (oncogene activation), the brakes cut (tumor suppressor loss), or both.
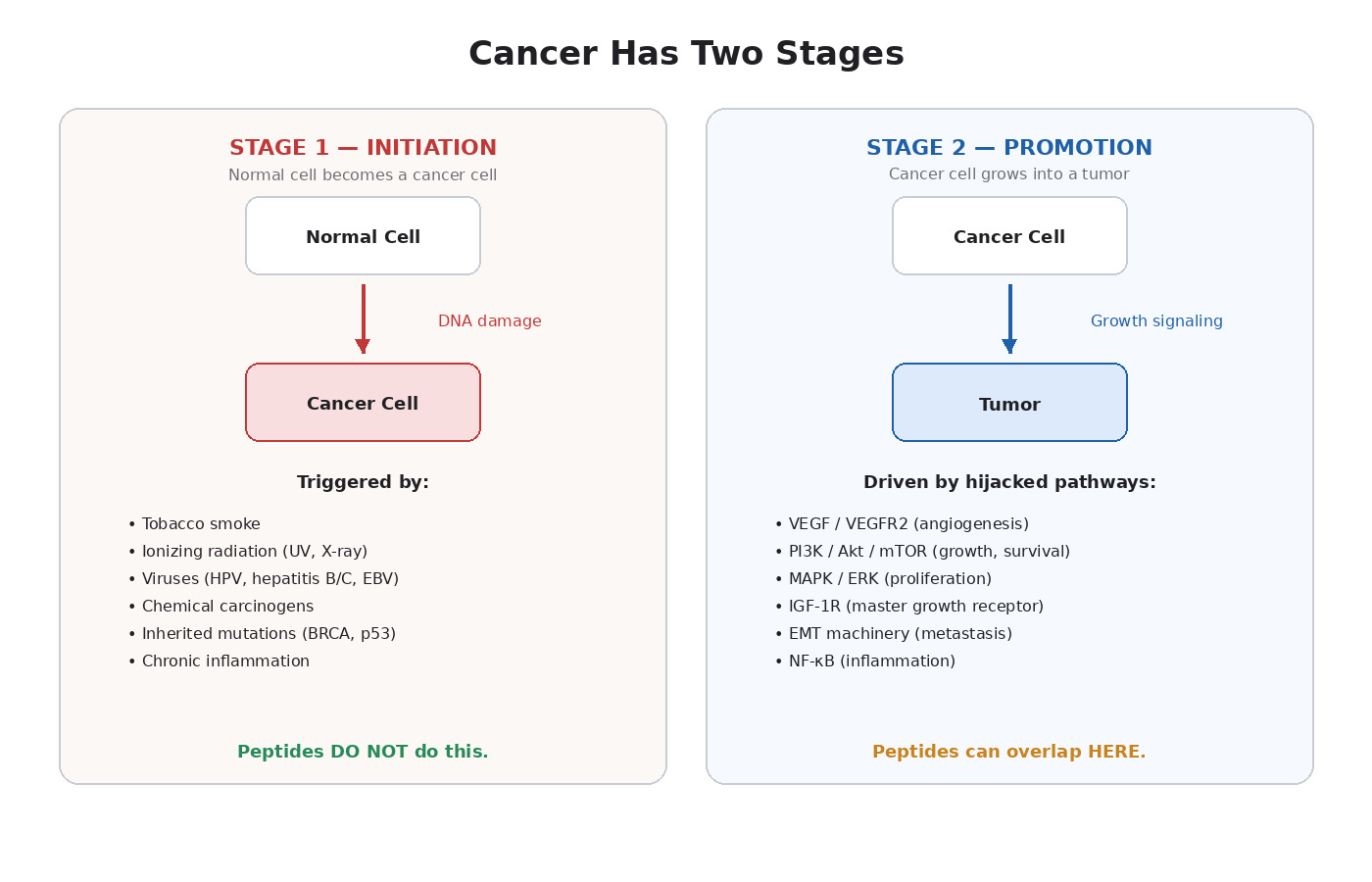
Stage One: Initiation
Initiation is the event that turns a normal cell into a cancer cell.
This requires real, physical damage to DNA — mutations, chromosome rearrangements, gene deletions, or changes that silence tumor suppressors. The well-mapped causes are short and specific:
- Tobacco smoke — damages DNA in lung, throat, bladder, and other tissue
- Ionizing radiation — UV light, X-rays, gamma rays. All of these can break DNA strands
- Certain viruses — HPV (cervical, head and neck), hepatitis B and C (liver), Epstein-Barr (some lymphomas)
- Chemical carcinogens — aflatoxins, asbestos, benzene, certain industrial chemicals
- Inherited mutations — BRCA1/2, Lynch syndrome genes, TP53 mutations (Li-Fraumeni)
- Chronic inflammation — long-running inflammation creates oxidative stress that damages DNA over time [1]
The key point: initiation requires DNA damage. Something has to break the DNA or permanently change how genes are read.
Peptides are not mutagens. None of the peptides this community discusses damage DNA. None of them activate oncogenes through mutation. None of them inactivate tumor suppressors through deletion. There is no published evidence that any of them transform a normal cell into a cancer cell.
This is the part most fear-based discussions of peptides skip entirely.
Stage Two: Promotion
Once a cancer cell exists, it doesn’t automatically become a tumor. A single mutated cell is fragile. To become dangerous, it has to:
- Survive — avoid being killed by the immune system or by its own internal alarm systems
- Grow — divide repeatedly to become a mass of cells
- Get blood supply — a tumor larger than about 1 to 2 millimeters cannot survive on diffusion alone
- Invade — break through the membranes that normally keep tissues separated
- Metastasize — travel to other organs and start new colonies
Each step requires signaling. Tumors don’t invent new biology. They hijack pathways your body already has — the ones used for normal healing, growth, and tissue maintenance [2].
This is where peptides enter the conversation.
The Pathways Tumors Hijack
A short preview of what later modules cover in depth:
- VEGF / VEGFR2 — used by healthy tissue for blood vessel formation during healing. Hijacked by tumors to build their own blood supply.
- PI3K / Akt / mTOR — used by healthy cells for growth and survival signaling. Hijacked by tumors to resist death and speed up division.
- MAPK / ERK — used for cell division in response to growth factors. Mutated or stuck on in roughly 30 percent of all cancers.
- IGF-1R — used for growth, especially in muscle and bone. Overexpressed in breast, prostate, colorectal, lung, and renal cancers [3].
- EMT machinery — used during embryo development and wound healing to let cells migrate. Hijacked by tumors to metastasize.
- NF-κB and inflammatory signaling — used by the immune system. Hijacked by tumors to create a pro-growth environment.
Several peptides this community uses work on these exact pathways. That is not because the peptides are oncogenic. It is because regeneration and tumor progression share machinery.
The Distinction in Plain Language
Imagine a building. Initiation is the act of someone setting a fire — a single event that turns a non-burning structure into a burning one. Promotion is everything that happens next: wind feeding the flames, dry wood letting the fire spread, accelerants making it burn hotter.
Peptides don’t set fires. But several of them can act like wind, or accelerant, if a fire already exists.
The takeaway is not “peptides are dangerous.” The takeaway is more specific: the risk profile of growth-signaling peptides depends almost entirely on whether a cancer cell already exists in the body. In a healthy person with no cancer, these compounds drive healing. In a person with an undetected tumor, they could in theory speed up its growth.
This is why family history, age, and baseline screening matter so much. We will get into all of that in later modules.
Key Takeaways
- Cancer biology has two stages: initiation (a normal cell becomes a cancer cell, requires DNA damage) and promotion (an existing cancer cell becomes a tumor, requires signaling).
- Peptides do not initiate cancer. They are not mutagens. They do not damage DNA.
- Several peptides work on pathways that tumors hijack during promotion. This is a real overlap, not a hypothetical one.
- The mechanism matters more than the molecule. A peptide that activates VEGFR2 in a healthy body drives healing. The same peptide in a body with an undetected tumor could feed angiogenesis.
- Risk is contextual, not universal. A 25-year-old with no family history and a clean screening profile is a different conversation than a 55-year-old with a first-degree relative who had hormone-sensitive cancer.
Next up: Module 2 — The Universal Cancer Playbook. What tumors actually do, and why every one of those behaviors maps to a pathway peptides interact with.
Module 2 — The Universal Cancer Playbook: Ten Tricks Every Tumor Uses
Research and educational purposes only. Not for human consumption.
Why This Module Matters
In 2000, two researchers named Hanahan and Weinberg published a paper called “The Hallmarks of Cancer” [4]. It became one of the most-cited papers in biology. Their argument: despite the huge variety of cancer types, every tumor acquires a small set of common abilities. Updates in 2011 [5] and 2022 [6] added more.
Once you understand the hallmarks, you understand cancer. And once you understand cancer, the peptide overlap stops being abstract. You can look at any compound and ask, “does this contribute to one of the hallmarks?”
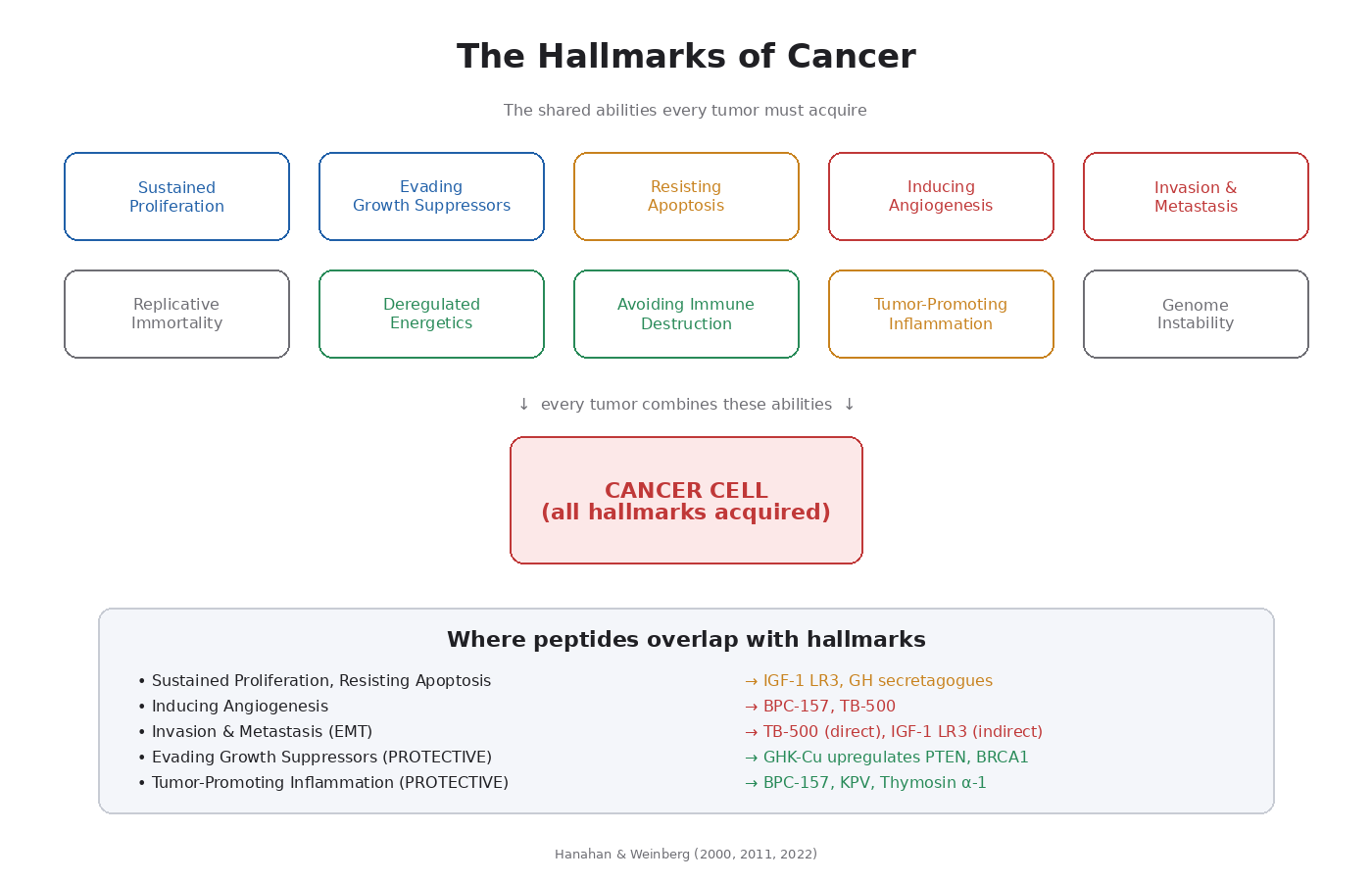
The Original Six Hallmarks
1. Sustained Proliferative Signaling
Normal cells need a signal from outside to divide. A growth factor arrives, binds to a receptor, and triggers an internal cascade that ends in cell division. Cancer cells bypass this. They either produce their own growth factors, multiply the number of receptors so they’re hyper-sensitive to any signal, or activate the downstream pathway so it’s always on.
Pathway most involved: PI3K/Akt/mTOR and MAPK/ERK. These are the two main proliferation cascades. We give each one its own module.
2. Evading Growth Suppressors
Normal cells have brakes. Tumor suppressor genes like p53, RB, and PTEN actively stop cells from dividing when they shouldn’t. Cancer cells either mutate these genes into non-working versions or shut down their expression.
Most famous suppressor: p53. It’s mutated in over 50 percent of all human cancers. People call it “the guardian of the genome” for a reason [7].
3. Resisting Cell Death (Apoptosis)
Every cell has a built-in self-destruct program. When DNA damage piles up, when growth signals go haywire, or when a cell finds itself in the wrong place, apoptosis is the safety net that takes it out. Cancer cells block this program. They overexpress anti-apoptotic proteins (like Bcl-2) or shut down pro-apoptotic ones (like Bax, Bak, and the caspase cascade).
This is one of the most important hallmarks for understanding peptide concerns. Compounds that broadly increase cellular survival signaling don’t just protect healthy cells. They protect malignant ones too.
4. Enabling Replicative Immortality
Normal cells can divide only a limited number of times before their telomeres (the protective caps on chromosomes) wear down and they stop dividing or die. Cancer cells reactivate telomerase, the enzyme that rebuilds telomeres, and gain unlimited replication.
Peptides generally don’t intersect with this hallmark directly. Epitalon, which the broader peptide community discusses, does upregulate telomerase. We touch on this in later modules.
5. Inducing Angiogenesis
A tumor larger than about 1 to 2 millimeters cannot survive on diffusion alone. The cells in the middle would starve. To grow beyond that size, tumors have to recruit new blood vessels. They do this by releasing VEGF (vascular endothelial growth factor), which tells nearby blood vessels to sprout new branches into the tumor [8].
This is the hallmark with the most direct peptide overlap. BPC-157, TB-500, and several others upregulate angiogenic signaling. Module 3 covers this in full.
6. Activating Invasion and Metastasis
The 90 percent of cancer deaths that aren’t from the primary tumor are from metastasis. Cells break off, travel through blood or lymph, and colonize distant organs. This requires a coordinated set of molecular changes called epithelial-mesenchymal transition (EMT), which we cover in Module 7.
The Updated Hallmarks (2011 and 2022)
Hanahan and Weinberg later added four more. All are relevant to the peptide conversation.
7. Deregulated Cellular Energetics
Cancer cells reprogram their metabolism. Most famously, they shift toward aerobic glycolysis (the Warburg effect). They burn glucose inefficiently even when oxygen is available, because the byproducts are useful for building new cells.
This is the angle that drugs like metformin attack. Metformin lowers glucose availability and disrupts mitochondrial complex I. Both are bad news for tumors.
8. Avoiding Immune Destruction
Your immune system actively patrols for cancer cells and kills most of them before they become tumors. Successful malignancies find ways to hide. They downregulate MHC molecules, express PD-L1 to shut down T-cells, or recruit regulatory T-cells that suppress immune responses.
Several peptides this community uses have immune effects (thymosin alpha-1, KPV, others). Most are immune-enhancing rather than suppressive. From the cancer-surveillance standpoint, that’s a favorable profile.
9. Tumor-Promoting Inflammation
Chronic inflammation is one of the strongest cancer drivers in the body. It creates oxidative stress (damaging DNA), supplies growth factors, and creates an environment that supports tumor cells.
This is why anti-inflammatory peptides — BPC-157 (in its anti-inflammatory mode), KPV, thymosin alpha-1 — have a more favorable risk profile than purely growth-signaling peptides.
10. Genome Instability and Mutation
Cancer cells accumulate mutations faster than normal cells. They have broken DNA repair, error-prone replication, and chromosome instability.
11. Unlocking Phenotypic Plasticity (added 2022)
Cancer cells can shift between differentiated and stem-cell-like states. That lets them resist treatment and adapt to new environments.
12. Senescent Cells (added 2022)
Senescent cells — old cells that have stopped dividing but haven’t died — release inflammatory signals that can drive tumor progression in nearby tissue.
Mapping the Hallmarks to Peptide Pathways
This is the central mapping to keep in mind for the rest of the course. Hallmark → driving pathway → peptide overlap:
- Sustained proliferative signaling → PI3K/Akt/mTOR, MAPK/ERK → IGF-1 LR3, growth hormone secretagogues
- Evading growth suppressors → p53, PTEN, RB → GHK-Cu actually upregulates some of these
- Resisting cell death → Bcl-2 family, caspases → IGF-1R signaling has anti-apoptotic effects
- Inducing angiogenesis → VEGF, VEGFR2 → BPC-157, TB-500
- Invasion and metastasis → EMT, MMPs, integrins → TB-500 (cell migration), IGF-1R
- Tumor-promoting inflammation → NF-κB, cytokines → most peptides are anti-inflammatory here
- Avoiding immune destruction → T-cell exhaustion, PD-L1 → thymosin alpha-1 enhances immunity
Notice the pattern. The growth and angiogenesis hallmarks are where peptide overlap concentrates. The immune and inflammation hallmarks are where many peptides are actually protective.
The Big-Picture Implication
Cancer is not a magic illness. It is a set of acquired abilities, each one built from normal cellular biology. Tumors don’t invent new pathways. They hijack the ones your body uses for growth, healing, and remodeling.
This is why the peptide conversation is more nuanced than “growth signal = cancer risk.” A peptide that activates a single receptor in a specific tissue type is very different from a peptide that broadly amplifies anti-apoptotic signaling across every cell.
Mechanism matters. The next several modules go pathway by pathway.
Key Takeaways
- Cancer has a small set of common abilities (hallmarks) that nearly every tumor acquires.
- Sustained proliferation, evading suppressors, resisting apoptosis, angiogenesis, invasion/metastasis, and replicative immortality are the original six.
- Inflammation, metabolic reprogramming, immune evasion, and genome instability were added later.
- Tumors don’t invent new pathways. They hijack normal ones.
- The peptide overlap concentrates on the growth and angiogenesis hallmarks.
- Some peptides are actually protective against inflammation and immune hallmarks.
- Mechanism specificity matters more than blanket categorization.
Next up: Module 3 — Feeding the Beast. The pathway most directly relevant to BPC-157 and TB-500.
Module 3 — Feeding the Beast: How Tumors Steal Your Blood Supply
Research and educational purposes only. Not for human consumption.
Why This Module Matters
Angiogenesis is the formation of new blood vessels. It is essential for wound healing, tissue regeneration, embryo development, and exercise adaptation. It is also one of the most directly hijacked processes in cancer biology. Roughly half of all human solid tumors depend on VEGF/VEGFR2 signaling to grow beyond a few millimeters [8].
This is the pathway BPC-157 most famously upregulates [9]. TB-500 also drives angiogenesis through related but distinct mechanisms. Understanding this pathway is non-negotiable for anyone using either compound.
The Basic Biology
Every cell in your body needs oxygen and nutrients. Blood vessels deliver those. The diffusion limit — the maximum distance oxygen can travel from a vessel into surrounding tissue — is roughly 100 to 200 micrometers. Any tissue thicker than that needs its own blood supply.
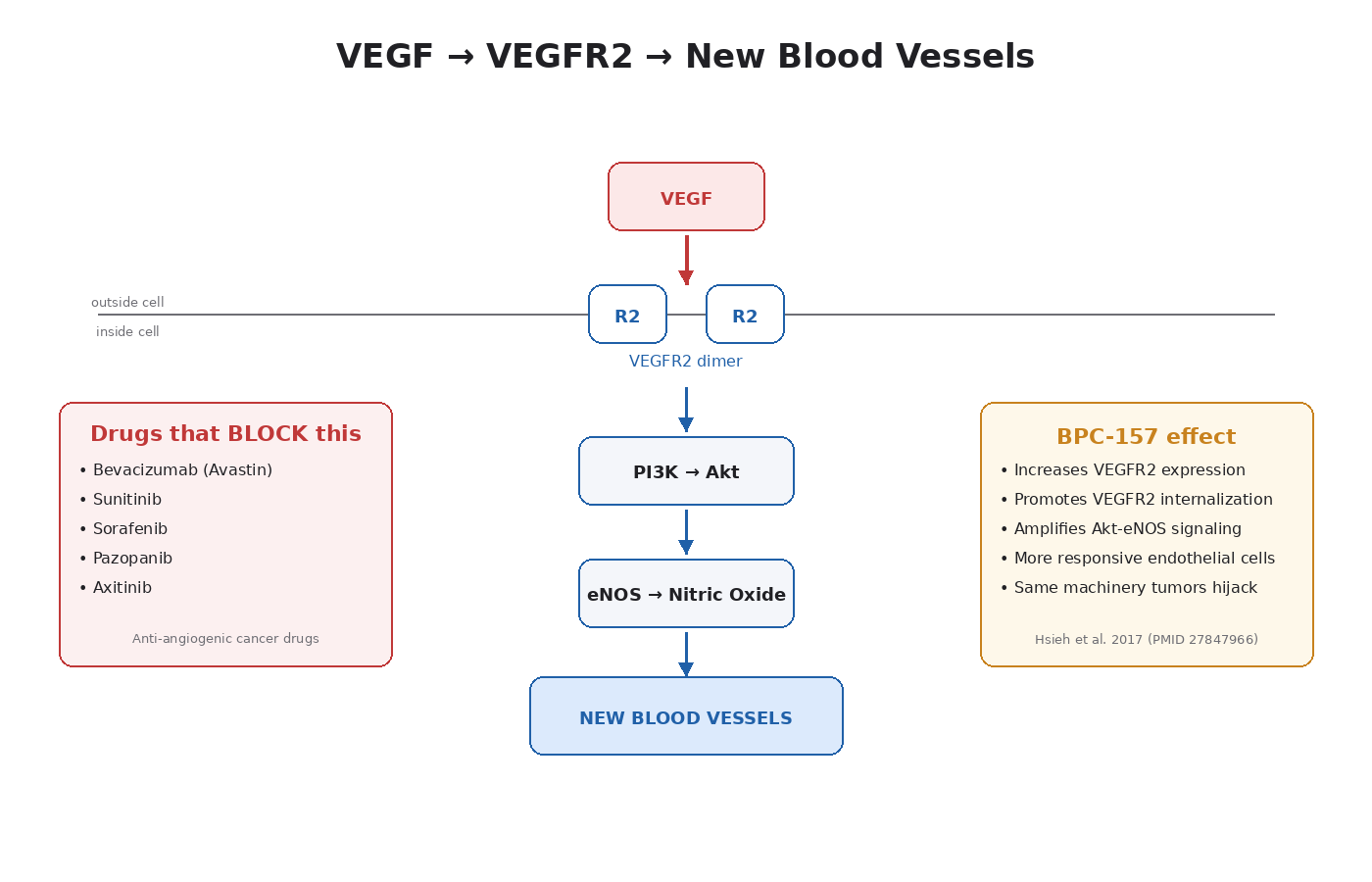
When tissue is damaged or hypoxic (oxygen-starved), it releases signals telling nearby blood vessels to sprout new branches into the affected area. The master signal is vascular endothelial growth factor (VEGF), a small protein that binds receptors on the endothelial cells lining existing blood vessels.
VEGF has several forms (VEGF-A, B, C, D, and placental growth factor), but VEGF-A is the dominant player in most contexts.
The receptor that matters most for blood vessel formation is VEGFR2 (also called KDR or Flk-1). When VEGF binds VEGFR2 on an endothelial cell:
- The receptor dimerizes (two copies pair up).
- The intracellular domains phosphorylate each other.
- This activates downstream pathways — mainly PI3K/Akt and PLC-γ/PKC.
- The endothelial cell starts dividing, migrating, and forming tube-like structures.
- New blood vessels sprout from existing ones.
The result is neovascularization — the formation of new capillaries that supply the previously underperfused tissue.
Why Tumors Need Angiogenesis
A microscopic cluster of cancer cells can survive on diffusion from nearby blood vessels. But as the cluster grows, the cells in the middle start to suffocate. Their oxygen sensors kick in. They release VEGF aggressively. Nearby blood vessels sprout new branches into the tumor.
This moment — when a tumor switches from being diffusion-dependent to being blood-supply-dependent — is called the angiogenic switch. It is one of the most studied events in tumor biology, and it is what separates harmless microscopic neoplasms from tumors that can grow to clinically meaningful sizes [8].
Tumors don’t use VEGF in normal amounts. They overproduce it, often dramatically. The new vessels they recruit are also abnormal: leaky, poorly organized, and weak. But they deliver enough oxygen and nutrients to let the tumor grow.
This is why anti-angiogenic drugs exist as a cancer treatment category. Bevacizumab (Avastin) is a monoclonal antibody that binds VEGF and prevents it from activating VEGFR2. It is approved for colorectal, lung, kidney, ovarian, and several other cancers. Sunitinib, sorafenib, pazopanib, and axitinib are small-molecule inhibitors of VEGFR2 used in renal cell carcinoma and other cancers.
The fact that an entire drug class exists to block what BPC-157 stimulates is why this question deserves serious attention.
BPC-157 and VEGFR2
The most rigorous mechanistic study of BPC-157’s angiogenic effect is Hsieh et al., published in 2017 [9]. The findings:
- BPC-157 increases vessel density both in vivo (in living animals) and in vitro (in cell culture).
- It speeds up blood flow recovery in models of hind limb ischemia (blocked blood supply).
- It increases VEGFR2 expression on vascular endothelial cells.
- It promotes the internalization of VEGFR2 — meaning the receptor gets pulled into the cell after activation, which is required for its full signaling effect.
- This drives activation of the VEGFR2-Akt-eNOS pathway, which produces nitric oxide and drives vasodilation and new vessel formation.
- All of these effects are blocked by dynasore, an inhibitor of endocytosis. This confirms that VEGFR2 internalization is the mechanism.
In plain language: BPC-157 makes endothelial cells more responsive to VEGF. It doesn’t necessarily increase VEGF levels themselves (the data on that is mixed), but it amplifies what VEGF does when it arrives.
For wound healing, this is excellent. For a body harboring an angiogenesis-dependent tumor, this is the same mechanism the tumor is already using.
The Counter-Evidence on BPC-157
This module would be dishonest if it didn’t include the other side of the literature.
In isolated human melanoma cell lines, BPC-157 has actually been shown to inhibit VEGF signaling via the MAPK kinase pathway. In a mouse C26 colon adenocarcinoma model, researchers giving BPC-157 saw reduced tumor cachexia (muscle wasting), preserved survival, and counteracted increases in pro-inflammatory cytokines like IL-6 and TNF-alpha — without speeding up tumor growth [10]. There are also unpublished mentions of reduced lung metastases in B-16 melanoma models, though “unpublished” carries less weight.
So the picture is not “BPC-157 always grows tumors.” The picture is “BPC-157 modulates angiogenic signaling, and the net effect appears to depend on context — driving healing in damaged tissue while showing some anti-tumor effects in specific cancer models.”
The honest read: we have very strong mechanistic data showing BPC-157 amplifies VEGFR2 signaling. We have limited and somewhat conflicting data on what that means in the context of an active cancer. The conservative position is to assume that broad upregulation of angiogenic machinery is not what you want happening in a body with undetected cancer cells.
TB-500 and a Different Angiogenic Mechanism
TB-500 (thymosin beta-4) drives angiogenesis too, but through a different mechanism:
- It directly stimulates endothelial cell migration and tube formation.
- It upregulates VEGF expression itself in a hypoxia-inducible factor (HIF-1alpha)-dependent way [11].
- It activates the FAK-paxillin pathway, which drives the cytoskeletal changes endothelial cells need to migrate.
- Its actin-sequestering function makes cell migration more efficient generally.
So TB-500 hits angiogenesis from the upstream side (more VEGF produced) while BPC-157 hits it from the receptor side (more responsive VEGFR2). Either way, you end up with more blood vessel formation.
Putting It Together — A Visual Walkthrough
Picture an injured tendon. Cells are damaged. Oxygen levels drop. Hypoxia-inducible factors activate. VEGF gets produced. Without BPC-157 or TB-500, this happens slowly. Endothelial cells eventually respond, capillaries sprout in, and the tissue heals over weeks.
Now picture the same injury with BPC-157. VEGFR2 expression goes up. The same amount of VEGF produces a much stronger response. Capillaries sprout faster. Blood flow restores faster. The tendon heals in less time.
Now picture a microscopic, undetected adenoma sitting somewhere — say, in the colon, where adenomatous polyps are common in adults over 40. That adenoma is at the edge of the angiogenic switch. It needs blood supply to grow. BPC-157 increases the responsiveness of nearby endothelial cells to the VEGF the adenoma is already producing.
The biology doesn’t care that you were targeting your tendon. The same machinery is operating throughout the body.
This is why baseline screening (colonoscopy at age-appropriate intervals, age-appropriate cancer screening in general) is part of the responsible-research framework for anyone using these compounds long-term.
Key Takeaways
- VEGF binds VEGFR2 to drive blood vessel formation. This is essential for healing.
- The same pathway is used by roughly half of all solid tumors to recruit blood supply.
- An entire class of cancer drugs (bevacizumab, sunitinib, sorafenib, others) exists to block VEGFR2 signaling.
- BPC-157 upregulates VEGFR2 expression and promotes its internalization, amplifying VEGF response.
- TB-500 increases VEGF production itself through HIF-1alpha and drives endothelial cell migration.
- BPC-157 has shown some anti-tumor effects in specific cancer models — the picture is more nuanced than blanket concern.
- The conservative position remains that broad amplification of angiogenic signaling is not ideal in a body with undetected cancer.
- This is why baseline screening matters, especially for users over 40 or with family history.
Next up: Module 4 — The Master Switch. The pathway most relevant to IGF-1 LR3.
Module 4 — The Master Switch: The Pathway Half of All Cancers Hijack
Research and educational purposes only. Not for human consumption.
Why This Module Matters
If you only learned one signaling pathway in all of biology, this would be the one to pick. PI3K/Akt/mTOR is the central decision-making system for whether a cell grows, divides, survives, or dies. It is involved in muscle growth, neuron survival, immune function, metabolism, longevity — and roughly 30 to 50 percent of all human cancers [12].
Every growth-signaling peptide in the research community feeds into this pathway. IGF-1 LR3 is the most direct activator. Growth hormone secretagogues activate it downstream through IGF-1. BPC-157 touches it through VEGFR2-Akt signaling. Insulin and IGF-1 both meet here.
This is the pathway you have to understand before any peptide discussion can be honest.
The Components, in Order
The full name is PI3K-Akt-mTOR. Each piece does something specific.
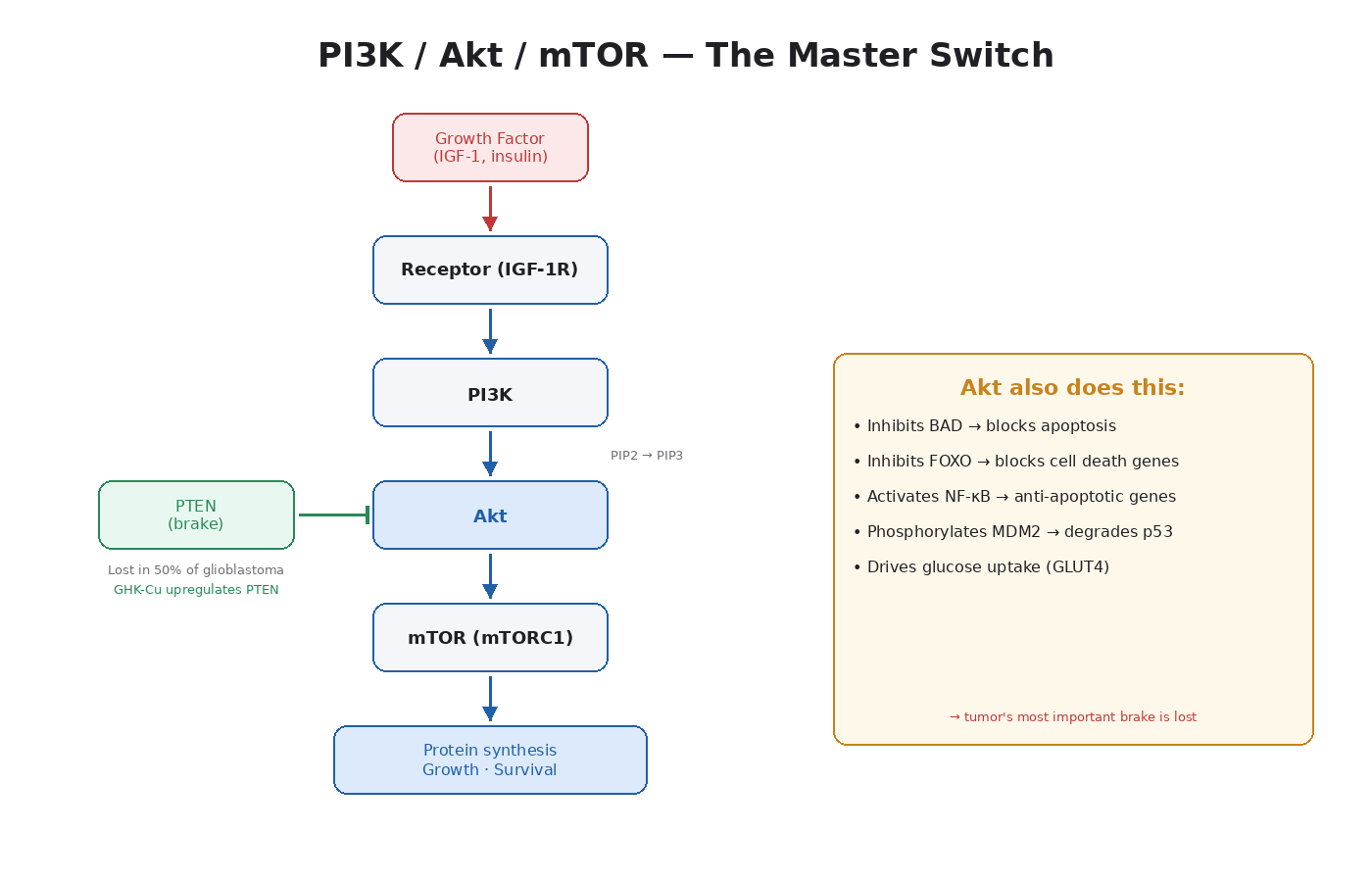
PI3K (Phosphoinositide 3-Kinase)
PI3K is an enzyme that sits just inside the cell membrane. When a growth factor binds its receptor on the outside (insulin to insulin receptor, IGF-1 to IGF-1R, etc.), the receptor activates PI3K. PI3K’s job is to take a membrane lipid called PIP2 and add a phosphate to it, turning it into PIP3.
PIP3 is the activating signal. Think of it as a beacon on the inside of the cell membrane that other proteins gather around.
Akt (also called PKB)
Akt is a protein kinase that gets pulled to the membrane by PIP3. Once there, it gets phosphorylated (activated) by two other kinases — PDK1 and mTORC2. Activated Akt then phosphorylates dozens of downstream targets.
Akt is the central node. It’s where a single input signal branches into a dozen different outputs. Activated Akt does all of the following:
- Activates mTOR (drives protein synthesis and cell growth)
- Inhibits FOXO transcription factors (which would otherwise activate cell death)
- Phosphorylates BAD (an apoptosis protein), preventing it from triggering cell death
- Activates GLUT4 glucose transporter movement to the membrane (insulin’s main metabolic effect)
- Inhibits GSK-3 (which would otherwise block glycogen synthesis)
- Phosphorylates MDM2, which then degrades p53 (the tumor suppressor)
That last one is important. Akt activation reduces p53 activity. A pathway that is upregulated by IGF-1 — and therefore by IGF-1 LR3 — actively suppresses the most important tumor suppressor in the human body.
mTOR (Mechanistic Target of Rapamycin)
mTOR is a serine/threonine kinase that comes in two complexes: mTORC1 (the most studied) and mTORC2.
mTORC1’s job is to integrate signals about whether the cell has enough resources to grow. It senses:
- Growth factor signaling (via Akt)
- Amino acid availability (especially leucine)
- Energy status (via AMPK)
- Oxygen availability
When all systems are go, mTORC1 activates two main downstream effectors:
- S6K1 — drives ribosome biogenesis and translation of growth-related proteins
- 4E-BP1 — releases the translation initiation factor eIF4E, accelerating protein synthesis
The net effect: the cell starts building proteins and growing.
mTOR is so central to growth that the drug rapamycin (which inhibits mTOR) is being studied as a longevity intervention. Chronic mTOR inhibition extends lifespan in essentially every model organism tested.
How This Pathway Drives Muscle Growth
This is the part the peptide community gets right intuitively.
When IGF-1 LR3 binds IGF-1R on a muscle cell:
- The receptor activates PI3K.
- PI3K converts PIP2 to PIP3.
- Akt gets pulled in and activated.
- Akt activates mTORC1.
- mTORC1 phosphorylates S6K1 and 4E-BP1.
- Protein synthesis ramps up.
- The muscle cell adds new contractile protein.
- Over time, this shows up as hypertrophy.
Resistance training does the same thing through mechanical stress that activates mTOR independently. Stack the two together and you get a multiplicative effect. That’s why IGF-1 LR3 has the reputation it does in athletic research.
How the Same Pathway Drives Cancer
Now swap “tumor cell” for “muscle cell” and the steps are identical:
- Growth factor (IGF-1, EGF, others) binds its receptor on a tumor cell.
- PI3K activates.
- Akt activates.
- mTORC1 activates.
- Protein synthesis ramps up.
- The tumor cell divides.
- Akt also blocks apoptosis — the cell can’t be killed easily.
- Akt phosphorylates MDM2 — p53 gets degraded — the tumor’s most important brake is lost.
- The tumor grows.
This is why PI3K/Akt/mTOR pathway activation is found in:
- ~40 percent of breast cancers
- ~30 percent of colorectal cancers
- ~50 percent of endometrial cancers
- ~60 percent of hepatocellular carcinomas
- A majority of glioblastomas
- Most prostate cancers [12]
Pathway components are commonly mutated. PIK3CA (which encodes a PI3K subunit) is one of the most frequently mutated oncogenes in human cancer. PTEN — the main brake on this pathway — is one of the most frequently lost tumor suppressors.
PTEN: The Brake You Want to Have
PTEN (Phosphatase and Tensin Homolog) is the enzyme that does the opposite of PI3K. It converts PIP3 back to PIP2, shutting down the pathway.
PTEN is one of the most important tumor suppressors in the human body. Loss of PTEN is found in:
- ~50 percent of glioblastomas
- ~30 to 70 percent of prostate cancers (depending on stage)
- ~30 percent of endometrial cancers
- A high percentage of metastatic melanoma
When PTEN is lost or inactivated, the PI3K/Akt/mTOR pathway becomes constitutively active — always on, no matter what’s happening upstream. This is one of the most dangerous configurations a cell can be in.
Notably, GHK-Cu has been shown to upregulate PTEN expression in human breast cancer (MCF7) and prostate cancer (PC3) cell lines [13]. This is one of the reasons GHK-Cu sits in a fundamentally different category than IGF-1 LR3, even though both are technically “peptides.”
The mTOR-IGF-1R Feedback Loop
This is a piece of biology with direct implications for cycling and washouts.
When mTOR is chronically active, it triggers a negative feedback loop that downregulates the insulin and IGF-1 receptors. This is the cell’s attempt to prevent runaway signaling. But there’s a flip side: when mTOR is inhibited (by drugs like rapamycin or temsirolimus), the feedback releases and IGF-1R signaling actually increases.
This is why mTOR inhibitors used as cancer drugs sometimes have limited effect as monotherapy — the tumor compensates by ramping up IGF-1R. Combination therapy with both mTOR inhibitors and IGF-1R inhibitors is being studied for exactly this reason.
The practical translation: chronic, uninterrupted growth signaling is a poor strategy, both for adaptation and for safety. Washouts let receptor density and signaling sensitivity reset.
Why This Pathway Is the Heart of the Course
Every other module in this course either feeds into or branches off from PI3K/Akt/mTOR.
- VEGFR2 (Module 3) activates this pathway as part of its angiogenic signaling.
- MAPK/ERK (Module 5) runs in parallel and crosstalks at multiple points.
- IGF-1R (Module 6) is the most direct upstream activator.
- p53 (Module 8) is degraded by Akt-activated MDM2.
- PTEN (Module 8) is the main brake on this pathway.
- Apoptosis resistance (Module 8) is largely mediated through Akt.
If you understand this pathway, you understand the molecular basis for almost every peptide-cancer concern in this space.
Key Takeaways
- PI3K/Akt/mTOR is the central growth and survival pathway in essentially every cell.
- Activated by growth factors (IGF-1, insulin, EGF), it drives protein synthesis, cell division, and apoptosis resistance.
- mTORC1 is the central integrator of “should this cell grow” signaling.
- Akt phosphorylates dozens of downstream targets, including MDM2 (which then degrades p53).
- The pathway is constitutively active or hyperactivated in 30 to 50 percent of all human cancers.
- PTEN is the main brake — its loss is one of the most dangerous oncogenic events.
- IGF-1 LR3 activates this pathway directly through IGF-1R.
- BPC-157 touches it through VEGFR2-Akt-eNOS.
- GHK-Cu actually upregulates PTEN, the brake on this pathway.
- Chronic uninterrupted signaling is unfavorable both for adaptation and for cancer risk — washouts matter.
Next up: Module 5 — The Most Broken Wire in Cancer. The parallel pathway, and the one with the most cancer mutations of any single cascade.
Module 5 — The Most Broken Wire in Cancer: RAS, RAF, and the Proliferation Engine
Research and educational purposes only. Not for human consumption.
Why This Module Matters
If PI3K/Akt/mTOR is the cell’s growth and survival pathway, MAPK/ERK is its proliferation pathway. Same upstream inputs in many cases (growth factor binding a receptor), different downstream outcome. Instead of “build proteins and resist death,” the message is “divide.”
Driver mutations in this pathway (specifically RAS) are the most common in all of cancer, appearing in roughly 30 percent of all tumor types [14]. BRAF mutations show up in about 8 percent. This is the pathway behind melanoma, pancreatic cancer, colorectal cancer, and a long list of other cancers.
IGF-1 LR3 activates MAPK/ERK in parallel with PI3K/Akt/mTOR. So does insulin, EGF, FGF, and most other growth factors. Understanding this pathway closes the loop on why growth-signaling peptides are categorically more cancer-relevant than other peptide categories.
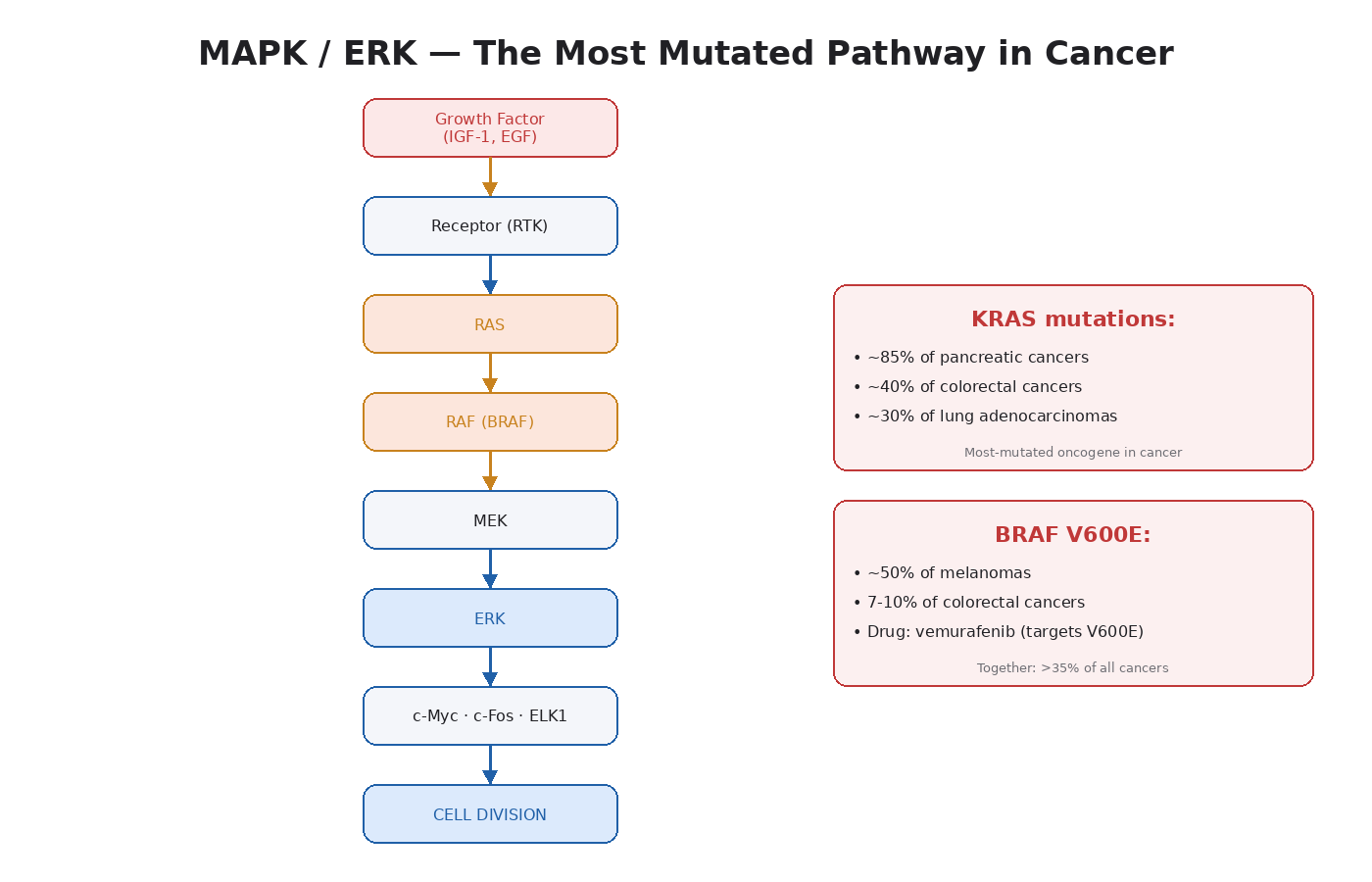
The Components, in Order
The full cascade is RTK → RAS → RAF → MEK → ERK. Each step is a kinase phosphorylating the next.
Receptor Tyrosine Kinase (RTK)
The starting point. A growth factor (IGF-1, EGF, FGF, PDGF, others) binds a receptor on the cell surface. The receptor dimerizes and the intracellular tyrosine residues get phosphorylated. This creates docking sites for adapter proteins.
GRB2 / SOS
GRB2 is an adapter protein that binds the phosphorylated receptor and recruits SOS, a guanine nucleotide exchange factor.
RAS
SOS swaps out the GDP on RAS for GTP. RAS-GTP is the active form. This is the central switch in the pathway. RAS activation triggers everything downstream.
There are three RAS proteins in humans: HRAS, KRAS, and NRAS. KRAS mutations are the most common driver mutation in all of cancer. They appear in ~85 percent of pancreatic cancers, ~40 percent of colorectal cancers, and ~30 percent of lung adenocarcinomas [14]. KRAS-G12D and KRAS-G12C are the specific point mutations that have become major drug targets in the last few years.
When RAS is mutated, it gets stuck in the GTP-bound active form. The pathway runs constantly.
RAF (ARAF, BRAF, CRAF)
Active RAS recruits and activates RAF kinases. RAF then phosphorylates the next kinase down.
BRAF V600E is the famous mutation here. It is found in roughly 50 percent of melanomas and 7 to 10 percent of colorectal cancers. The drug vemurafenib was developed specifically to inhibit BRAF V600E and changed melanoma treatment when it was approved.
MEK1 / MEK2
RAF phosphorylates MEK, which then phosphorylates ERK. MEK mutations are rare in cancer, but MEK inhibitors (trametinib, cobimetinib) are used clinically to block hyperactivated MAPK signaling downstream of RAS and BRAF mutations.
ERK1 / ERK2
The end of the cascade. Phosphorylated ERK moves into the nucleus and activates transcription factors — ELK1, c-Fos, c-Myc, c-Jun. These transcription factors drive expression of genes that promote cell division.
The output: the cell enters the cell cycle and divides.
How This Pathway Drives Normal Cell Division
In a healthy context, this cascade is tightly regulated. A growth factor arrives. The cascade fires briefly. The cell divides. Then negative feedback loops shut everything off. Phosphatases dephosphorylate ERK. DUSPs (dual specificity phosphatases) get expressed and clean up phosphorylated components. The system resets.
This is how wound healing, immune response, and tissue maintenance work. Growth signals are pulsatile and brief.
How the Same Pathway Drives Cancer
In a tumor cell, one of two things is broken:
Constitutive activation — a mutation (KRAS, BRAF, NRAS) keeps the pathway permanently on. The cell divides whether or not external growth signals are present.
Hyperresponsiveness — receptor overexpression (HER2 amplification in breast cancer, EGFR amplification in lung cancer) means the cell responds dramatically to even tiny amounts of growth factor.
Either way, ERK is chronically active. The transcription factors it drives — especially c-Myc — turn on a massive program of cell division genes. The cell divides over and over.
ERK also drives:
- Invasion and metastasis through matrix metalloproteinase expression
- Angiogenesis through VEGF expression
- Anti-apoptotic signaling through Bcl-2 family modulation
So MAPK/ERK doesn’t just drive proliferation. It hits multiple hallmarks of cancer at once.
How Peptides Touch This Pathway
This is where the peptide overlap shows up:
IGF-1 LR3 activates MAPK/ERK directly. When IGF-1 binds IGF-1R, the receptor activates both PI3K (Module 4) and the RAS-RAF-MEK-ERK cascade in parallel. This dual activation is why IGF-1 is such a potent growth signal. It hits both pathways at the same time.
Growth hormone secretagogues (CJC-1295, ipamorelin, MK-677, hexarelin, others) activate this pathway indirectly through endogenous IGF-1 production.
BPC-157 has been shown to activate MAPK signaling in some studies. In human melanoma cell lines it actually inhibited VEGF signaling via MAPK — a context-dependent effect.
TB-500 drives MAPK activation in the context of cell migration and wound healing.
EGF, FGF, and PDGF analogs (less common in the research peptide community but worth mentioning) all activate MAPK/ERK as their primary effect.
The Crosstalk Problem
PI3K/Akt/mTOR (Module 4) and MAPK/ERK don’t operate alone. They cross-regulate each other at multiple points:
- RAS activates both pathways simultaneously.
- Akt can phosphorylate RAF (sometimes positively, sometimes negatively).
- ERK can phosphorylate TSC2, indirectly activating mTOR.
- mTOR feedback can dampen IGF-1R, which dampens both pathways.
This crosstalk is why blocking just one pathway in cancer treatment often fails — the other compensates. Combination therapies (MEK inhibitor + PI3K inhibitor) are being explored for exactly this reason.
The practical implication for peptide users: when you activate IGF-1R with IGF-1 LR3 or push endogenous IGF-1 up with growth hormone secretagogues, you are activating both PI3K/Akt/mTOR and MAPK/ERK in parallel. That dual activation is what makes the signal so anabolic — and it is also what makes the cancer overlap more concerning than activating either pathway alone.
Why This Pathway Is Where Mutations Concentrate
RAS proteins are mutated in ~30 percent of all human cancers. BRAF in ~8 percent. Combined, mutations in RAS-RAF-MEK-ERK pathway components are present in well over a third of all cancers.
Why this pathway specifically?
- RAS sits at a node where many growth factor receptors converge. A mutation here affects multiple inputs at once.
- The pathway is short — only four or five steps — so a single mutation can have an outsized effect.
- The downstream outputs (proliferation, anti-apoptosis, angiogenesis) hit multiple cancer hallmarks at once.
- RAS proteins have historically been “undruggable” — no good binding pocket — which is why tumors with these mutations have been so hard to treat. (KRAS-G12C inhibitors like sotorasib have changed this in the last few years.)
When you understand that this is the most mutated pathway in all of cancer, you understand why activating it (even therapeutically, even in a healthy context) deserves serious thought about cycling and risk stratification.
Key Takeaways
- MAPK/ERK is the central proliferation pathway in essentially every cell.
- The cascade is RTK → RAS → RAF → MEK → ERK.
- Driver mutations in this pathway (especially KRAS and BRAF) appear in roughly a third of all cancers.
- ERK drives cell division through transcription factors like c-Myc, c-Fos, ELK1.
- It also drives angiogenesis, invasion, and apoptosis resistance — multiple hallmarks at once.
- IGF-1 LR3 activates this pathway in parallel with PI3K/Akt/mTOR — dual activation amplifies anabolic effect and risk.
- Crosstalk between MAPK/ERK and PI3K/Akt/mTOR means activating one effectively activates both.
- This is the most mutated pathway in all of human cancer, which is why activating it deserves serious thought about cycling.
Next up: Module 6 — The Receptor That Sits at the Center of Everything.
Module 6 — The Receptor That Sits at the Center of Everything
Research and educational purposes only. Not for human consumption.
Why This Module Matters
The IGF-1 receptor (IGF-1R) sits at the crossroads of everything covered in the last two modules. It is the most direct upstream activator of both PI3K/Akt/mTOR and MAPK/ERK. It is overexpressed in nearly every major solid tumor type. Chronically elevated circulating IGF-1 is one of the most consistent epidemiological signals for cancer risk in the entire scientific literature [3].
This is also the pathway with the most direct and documented overlap with peptides this community uses. IGF-1 LR3 is a synthetic IGF-1 analog with a longer half-life and weaker binding to IGF-binding proteins. That means it activates IGF-1R more efficiently and for longer than native IGF-1. Growth hormone secretagogues (CJC-1295, ipamorelin, MK-677, tesamorelin) raise endogenous IGF-1 levels through GH-mediated liver production.
If you are using any compound that pushes IGF-1 signaling, you have to understand this module.
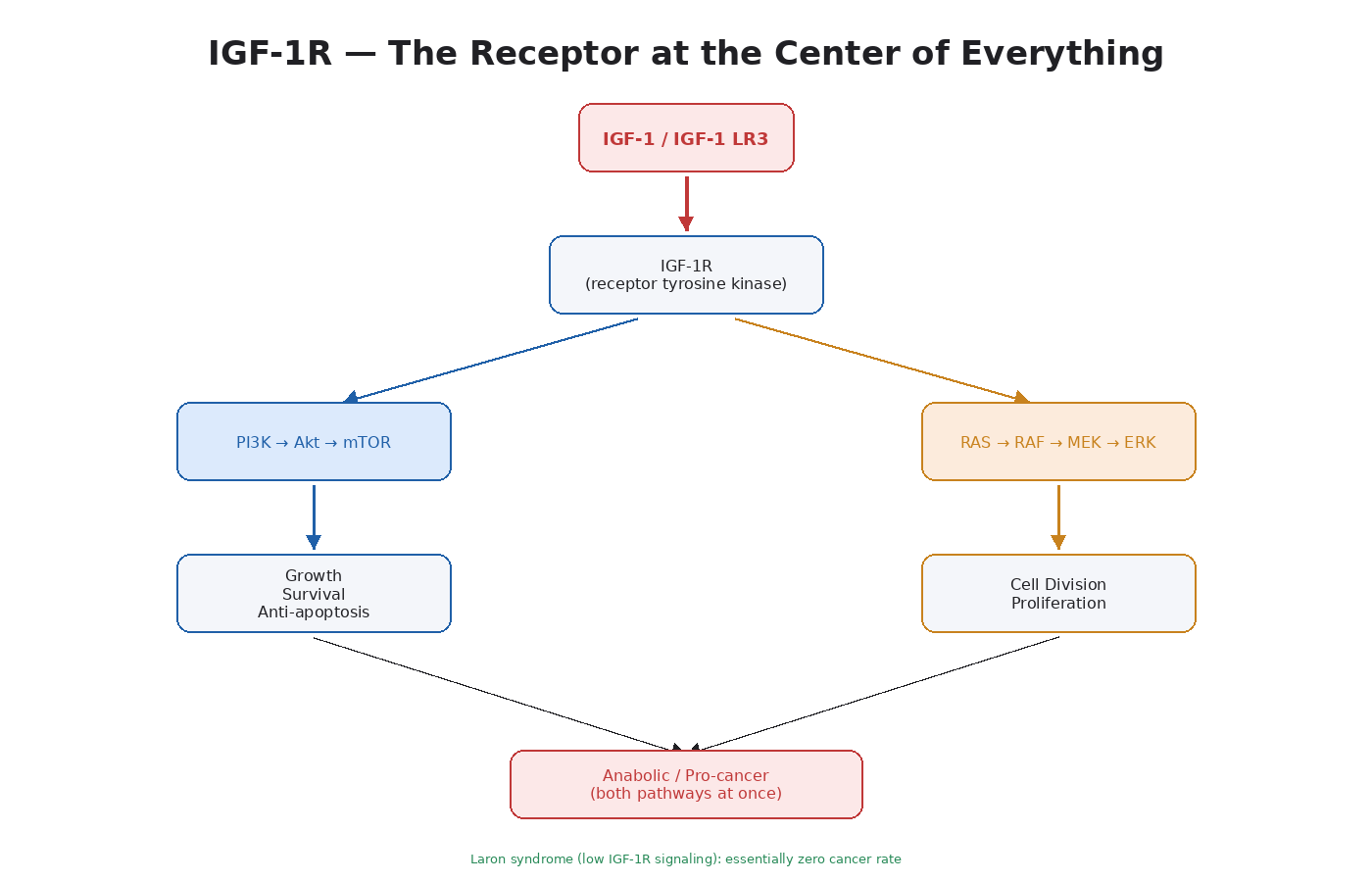
The Basic Biology of IGF-1R
The insulin-like growth factor 1 receptor is a tyrosine kinase receptor that sits on the surface of nearly every cell in the body. It is structurally very similar to the insulin receptor — so similar that the two can form hybrid receptors when both are expressed in the same cell.
When IGF-1 binds IGF-1R:
- The receptor dimerizes and autophosphorylates.
- This recruits IRS-1 (insulin receptor substrate 1) and Shc adapter proteins.
- IRS-1 activates PI3K → Akt → mTOR (Module 4).
- Shc activates RAS → RAF → MEK → ERK (Module 5).
- Both pathways fire at the same time.
The dual activation is what makes IGF-1 such a potent growth signal. It hits the survival/protein synthesis pathway and the proliferation pathway at once.
What IGF-1 Does in Healthy Tissue
IGF-1 is produced mainly by the liver in response to growth hormone signaling. It also has local (paracrine) production in tissues where growth is happening — muscle, bone, brain, gut.
The major effects:
- Muscle growth — activates satellite cells, drives protein synthesis, promotes hypertrophy
- Bone growth — drives chondrocyte proliferation and bone elongation during childhood
- Neuronal survival — one of the most important neuroprotective growth factors in the brain
- Tissue regeneration — supports healing across most organ systems
- Glucose homeostasis — overlaps with insulin signaling at the metabolic level
- Anti-apoptotic protection — keeps cells alive in conditions that would otherwise trigger cell death
This is all good in a healthy body. IGF-1 deficiency causes growth failure in children and is linked to frailty in older adults. People with naturally low IGF-1 (like individuals with Laron syndrome) have very specific health profiles.
What IGF-1 Does in a Body with Cancer
The same effects, in tumor cells, are catastrophic.
- Anti-apoptotic protection means cancer cells that should die, don’t.
- Pro-proliferative signaling means they divide faster.
- Survival signaling protects them from chemotherapy and radiation.
- The PI3K/Akt branch contributes to angiogenesis through HIF-1alpha stabilization.
The epidemiological data on this is substantial:
- Elevated circulating IGF-1 is associated with increased risk of colorectal, breast, and prostate cancer in multiple large cohort studies [3].
- People in the highest quartile of IGF-1 have measurably higher rates of certain cancers compared to those in the lowest.
- Laron syndrome patients (who have IGF-1R signaling deficiencies) have an essentially zero cancer rate in the populations studied. The famous Ecuadorian cohort showed dramatically reduced cancer mortality [15].
- Acromegaly patients (who have chronically elevated IGF-1 from GH-secreting pituitary tumors) have higher rates of colorectal cancer specifically.
The biological explanation is that IGF-1R is overexpressed or hyperactive in:
- Breast cancer
- Prostate cancer (both androgen-dependent and androgen-independent forms)
- Colorectal cancer
- Lung cancer
- Renal cell carcinoma (where IGF-1R overexpression carries a 70 percent increased death risk in clear cell RCC)
- Hepatocellular carcinoma
- Adrenocortical carcinoma
- Many sarcomas (especially Ewing sarcoma)
IGF-1R has been a major drug development target for over a decade. Several monoclonal antibodies (figitumumab, ganitumab) and small-molecule inhibitors (linsitinib, BMS-754807) have been developed specifically to block IGF-1R in cancer treatment.
IGF-1 LR3 Specifically
IGF-1 LR3 is a synthetic analog of IGF-1 with two key modifications:
- R3 substitution — the third amino acid is changed from glutamic acid to arginine. This dramatically reduces binding to IGF-binding proteins (IGFBPs).
- N-terminal extension — 13 extra amino acids on the front end, which further reduces IGFBP binding.
The result is a peptide with a much longer half-life than native IGF-1 (roughly 20 to 30 hours vs ~10 minutes) and 2 to 3 times the bioavailability at the IGF-1 receptor.
In normal physiology, IGFBPs (especially IGFBP-3) bind most of the IGF-1 in circulation. Only a small fraction is free to bind IGF-1R at any given time. This is a built-in safety mechanism — it keeps IGF-1 signaling tightly regulated.
IGF-1 LR3 bypasses this regulation. It binds IGF-1R freely, stays in circulation longer, and produces a sustained, elevated signal that doesn’t occur in normal biology.
This is exactly what makes it anabolic. It’s also what makes it the peptide with the most direct overlap with documented cancer-promoting signaling.
The Insulin Receptor Overlap
IGF-1R and the insulin receptor are roughly 60 percent identical at the amino acid level. They can form hybrid receptors. They activate overlapping downstream pathways.
This matters because:
- IGF-1 has weak insulin-like effects on glucose disposal at high doses.
- Chronic IGF-1 signaling can contribute to insulin resistance.
- Insulin itself can bind IGF-1R at high concentrations (this is why hyperinsulinemia from poorly controlled type 2 diabetes is associated with cancer risk).
- The hybrid receptors complicate the signaling — they’re more sensitive to IGF-1 than to insulin.
The cancer-relevant consequence: chronic hyperinsulinemia and chronic IGF-1 elevation work on overlapping receptors and overlapping pathways. Both have been linked to increased cancer risk, especially for colorectal, breast, and endometrial cancers. The shared underlying mechanism is sustained PI3K/Akt/mTOR activation.
IGFBPs: The Built-In Brake System
IGF-binding proteins (IGFBPs 1 through 6) are a family of proteins that bind IGF-1 and IGF-2 in circulation. They serve several functions:
- Reservoir — sequester IGF-1 so it doesn’t degrade quickly.
- Modulator — control how much free IGF-1 is available to bind IGF-1R.
- Independent effects — some IGFBPs (especially IGFBP-3) have IGF-independent pro-apoptotic effects.
IGFBP-3 specifically is interesting because it has direct anti-cancer effects independent of IGF-1 binding. It can induce apoptosis in tumor cells through interactions with the nuclear receptor RXR-alpha. Some research even suggests that the ratio of IGF-1 to IGFBP-3 may be a better predictor of cancer risk than absolute IGF-1 levels.
IGF-1 LR3 bypasses IGFBPs completely. It does not engage this regulatory layer. This is mechanistically why it’s so much more potent than native IGF-1 — and why its long-term safety profile is harder to characterize.
What This Means for Growth Hormone Secretagogues
Compounds like CJC-1295, ipamorelin, MK-677, and tesamorelin don’t directly bind IGF-1R. They raise endogenous IGF-1 through the GH axis:
- The peptide stimulates pituitary GH release (or in the case of GHRP-class compounds, both GH release and increased pulsatility).
- GH binds GH receptors on the liver.
- The liver produces IGF-1.
- IGF-1 circulates and binds IGF-1R throughout the body.
The signaling endpoint is the same as injecting IGF-1, but the kinetics are different. Endogenous IGF-1 is regulated by IGFBPs, has natural diurnal variation, and is subject to the body’s feedback systems. Pulsatile GH release (which is what natural physiology does) produces pulsatile IGF-1, which is biologically different from sustained elevation.
This is why the cancer-risk profile of well-cycled growth hormone secretagogues is generally considered more favorable than chronic IGF-1 LR3 use, even though the downstream pathways being activated are similar. Module 13 covers this in more detail.
Risk Stratification, Briefly
A few high-risk profiles for IGF-1-mediated peptides:
- Personal history of any cancer — particularly breast, prostate, colorectal, or endometrial
- First-degree family history of these cancers
- BRCA1/2 mutations or other inherited cancer syndromes
- Active or treated diabetes with hyperinsulinemia
- Age above ~50 where subclinical neoplasms become statistically more likely
- Acromegaly or any condition with baseline elevated IGF-1
- Hormone-sensitive cancers in family — these have particularly strong IGF-1R coupling
This isn’t a checklist for permission. It’s a checklist for taking the risk discussion seriously.
Key Takeaways
- IGF-1R is the receptor that sits at the intersection of PI3K/Akt/mTOR and MAPK/ERK.
- It is overexpressed or hyperactive in nearly every major solid tumor type.
- Chronically elevated circulating IGF-1 is one of the most consistent cancer risk signals in epidemiology.
- Laron syndrome patients (low IGF-1 signaling) have dramatically reduced cancer rates.
- IGF-1 LR3 bypasses the IGFBP regulatory system, producing sustained signaling that doesn’t occur naturally.
- The insulin receptor and IGF-1R overlap structurally and functionally — chronic hyperinsulinemia hits the same pathways.
- Growth hormone secretagogues raise IGF-1 indirectly and pulsatile-ly, which is biologically different from direct LR3 injection.
- Risk stratification matters more here than for any other peptide category in this course.
Next up: Module 7 — The Migration Switch. The pathway behind metastasis, and the most direct overlap point for TB-500.
References
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–867. PMID: 12490959.
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. PMID: 10647931.
- Pollak MN. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8(12):915–928. PMID: 19029956.
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. PMID: 10647931.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. PMID: 21376230.
- Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12(1):31–46. PMID: 35022204.
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–310. PMID: 11099028.
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307. PMID: 21593862.
- Hsieh MJ, Liu HT, Wang CN, et al. Therapeutic potential of pro-angiogenic BPC157 is associated with VEGFR2 activation and up-regulation. J Mol Med. 2017;95(3):323–333. PMID: 27847966.
- Sikiric P, Seiwerth S, Rucman R, et al. Stable gastric pentadecapeptide BPC 157: novel therapy in gastrointestinal tract. Curr Pharm Des. 2011;17(16):1612–1632. PMID: 21548867.
- Smart N, Risebro CA, Melville AAD, et al. Thymosin β4 induces adult epicardial progenitor mobilization and neovascularization. Nature. 2007;445(7124):177–182. PMID: 17108969.
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase-AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. PMID: 12094235.
- Pickart L, Margolina A. Regenerative and protective actions of the GHK-Cu peptide in the light of the new gene data. Int J Mol Sci. 2018;19(7):1987. PMID: 29986520.
- Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26(22):3291–3310. PMID: 17496923.
- Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med. 2011;3(70):70ra13. PMID: 21325617.
- Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. PMID: 19945376.
- Sribenja S, Wongkham S, Wongkham C, Yao Q, Chen C. Roles and mechanisms of beta-thymosins in cell migration and cancer metastasis: an update. Cancer Invest. 2013;31(2):103–110. PMID: 23320793.
Continue the series:
→ Part 2: Metastasis, Tumor Brakes, and the First Peptide Deep Dives